
记忆B细胞传统意义上被定义为生发中心(GC)B细胞的后代,表达同型转换和突变后的B细胞受体(BCRs)。然而,这不是记忆B细胞的唯一定义。我们在这里讨论小鼠的记忆B细胞,根据以下几个方面进行定义:(1)细胞表面标记;(2)多层级;(3)以T细胞依赖及GC依赖/非依赖的方式形成;(4)以不依赖T细胞的方式形成;(5)有自身免疫性疾病的小鼠模型。
未成熟B细胞表面表达BCR,这是一种与信号分子Igα/β相关的膜结合抗体。B细胞在骨髓中发育完成后,未成熟B细胞通过血液迁移到二级(外周)淋巴器官,如脾脏,在那里分化为成熟的B细胞。成人中,B细胞的发育在骨髓中进行,产生B1和B2 B细胞亚群。B1可以进一步细分为B1a和B1b,其中大部分B1a B细胞来自胎儿肝脏,而B2细胞分为滤泡(FO)和边缘区(MZ)B细胞。B1和MZ B细胞是天然抗体的来源,对非T细胞依赖(Ti)抗原起免疫反应。血液、脾脏和淋巴结中的主要亚群是FO B细胞,主要对T细胞依赖(Td)抗原做出反应。在B细胞被激活后,它们可以分化为记忆细胞和/或分泌抗体的浆细胞。
经过激活后,FO B细胞与滤泡树突状细胞(FDCs)和滤泡T辅助细胞(TFH)一起形成生发中心(GC),这是位于B细胞滤泡内的次级结构。FDCs以免疫复合物的形式捕获并保留抗原在其表面,TFH细胞被发现通过同源相互作用为B细胞提供分化信号。
GCs还支持BCR修饰,即类别转换重组(CSR)和体细胞超突变(SHM),这些过程需要激活诱导脱氨酶(AID)。GC可分为两个区,一个是B细胞进行克隆性扩增的暗区;一个是B细胞根据其与FDCs和T辅助细胞相互作用的能力进行选择的明区。当B细胞离开GCs时,它们分化为记忆B细胞或产生抗体的浆细胞,由于SHM和/或CSR导致效应功能的改变,表达的BCRs可能会经历亲和力成熟。
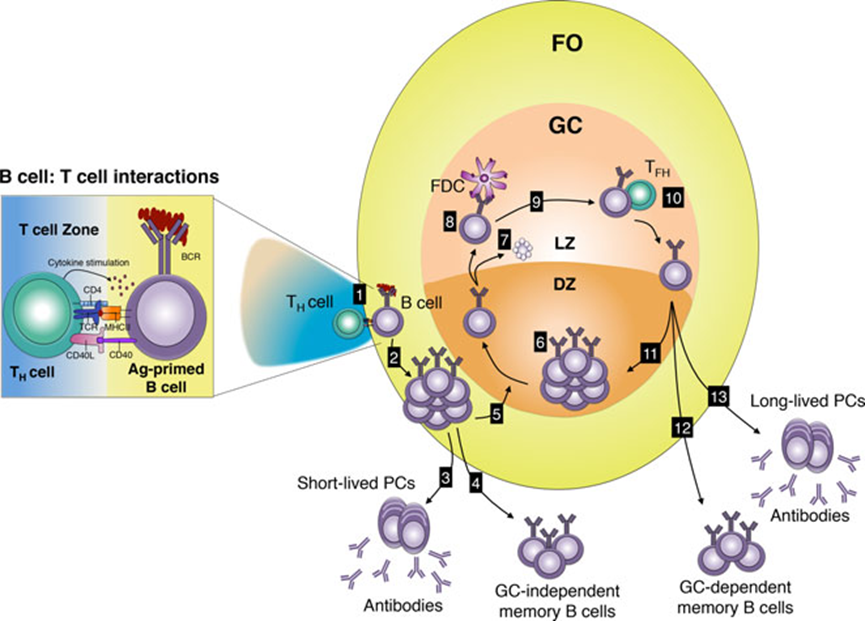
图1 记忆B细胞形成途径示意图
在人体中,记忆B细胞的比例远远高于小鼠,至少无病原体条件下的。并且人体记忆B细胞的主要特征是表达CD27,这是一种有抗原经历细胞的标志。在人类的CD27+B细胞中,存在着IgM和经历过SHM的同型转换的细胞。此外,缺乏CD27表达的记忆B细胞也有报道。有观察显示,CD27并不是小鼠记忆B细胞的适当标志物,并且由于记忆B细胞的数量少,在技术上难以仔细研究它们。为了避免这个问题,许多研究都依赖于使用杂交瘤和表达特定抗体的H链,单独或与确定的L链结合的转基因小鼠(TG),产生的B细胞可以高频率表达具有预先确定的抗原特异性的BCR。将此类结构体引入Ig的H(和L)链位点,也可以实现CSR,因此,研究可以表达同型转换抗原特异性BCRs的B细胞是可能的。
传统定义下,记忆B细胞为表达同型转换和大量突变BCRs的GC B细胞的后代。因此,对记忆B细胞的研究是针对表达同型转换抗原特异性BCR的B细胞,这些B细胞在GC反应停止后很久还存在。然而,最近在追踪记忆B细胞方面的进展使彻底研究这些细胞的性质成为可能,甚至是在不使用TG BCR的情况下。这揭示了记忆B细胞池在其生成、分化、功能、表型标记以及其BCR的SHM和/或同型转换水平方面具有未曾预见的异质性。此外,有证据表明还有一些途径可以形成记忆B细胞,这是一条Td但与GC无关的途径。不仅如此,甚至针对T细胞无关抗原,记忆B细胞也能形成。
下面根据以下几个方面讨论各种记忆B细胞群的定义:(1)细胞表面标记;(2)多层级;(3)以T细胞依赖性以及GC依赖性或非依赖性的方式形成;(4)以不依赖T细胞的方式形成;(5)有自身免疫性疾病的小鼠模型。
使用抗原特异性记忆B细胞的相对数量较高的TG小鼠模型,通过对细胞表面标志物进行研究,使记忆B细胞的定义得以确定,并且这在非TG小鼠中得到了证实。在这个系统中,B细胞表达一个明确的H链,与内源性λ1 L链结合,形成针对半抗原4-羟基-3-硝基苯乙酰 (NP)的特异性BCR。在这项研究中,用NP与鸡γ球蛋白(CGG)进行免疫后,刺激因子CD80被确定为记忆B细胞的标志物。可以与NP结合的IgM和IgG记忆B细胞被发现,并且其中60%以上表达CD80,其中大多数(70%)经过SHM。这也意味着在同型转换的记忆B细胞中,存在着表达非突变BCR的细胞,因此与传统的记忆B细胞的观点相悖。
后来CD80结合CD73和PD-L2两种标志物,区分了至少五种针对Td抗原NP-CGG免疫反应的记忆B细胞的亚群。由于在这五个亚群中都检测到了IgM和同型转换的细胞,说明亚群分类与同种型的表达无法关联。这些数据表明,记忆B细胞群的多样性是相当大的,存在一个谱系。这个谱系将由原生的记忆B细胞和其他更多的类记忆细胞组成,前者BCR不常进行同型转换且很少发生突变,而后者表达多种特异性标志物,更频繁地进行同型转换产生BCR且高突变含量高。

图2 B细胞的五种亚群的区分
在另一个模型系统中,使用黄色荧光蛋白(YFP)标记表达AID的细胞。其假设是,SHM和CSR所需的AID在GC反应期间被激活,因此YFP不仅会标记GC B细胞,也会标记它们的后代。这个模型允许长时间追踪YFP阳性细胞针对一种微粒状Td抗原的羊红血球细胞(SRBC)或可溶性Td抗原的NP-CGG所产生的免疫反应。通过这种方法,发现在SRBC免疫后长达8-12个月,都能检测到IgM和IgG记忆B细胞,而经过NP-CGG免疫后,只在3-4个月内可以检测到这些细胞,这表明对微粒抗原的反应产生的记忆会更持久,抗原的性质对记忆B细胞反应的持续时间很重要。此外,IgM记忆B细胞也有会继续分化。
对四个不同的YFP阳性记忆B细胞亚群以细胞表面标志物的方式进行区分。这些细胞可以根据IgM和IgG表达情况,以及它们是否与花生凝集素(PNA)结合进行划分。尽管所有的亚群都显示出了SHM的迹象,但无论哪种亚型,PNA阳性部分的频率都较高,并随时间而变化。此外,PNA阳性和PNA阴性的部分都是CD73和CD80阳性,只是它们的Fas(CD95)表达水平不同。记忆B细胞上CD73和CD80的表达情况与上文(1)讨论的记忆B细胞标志物一致。PNA和Fas也是GC B细胞的标志物,与此相一致的是,在SRBC免疫后的8个月内都可以检测到类似GC的结构。PNA+细胞的存在和GC反应开启了记忆B细胞再循环的可能性。事实上,IgM和IgG记忆亚群的过继转移显示,前者产生了GCs,而后者则分化为浆细胞,这也表明了记忆B细胞亚群的不同功能。由于AID的表达也可以发生在GC结构之外,YFP的阳性可能不是那些通过GC的细胞所独有的。尽管如此,这些数据与以前所认识到的更具可塑性和异质性的记忆B细胞反应相一致。基于这些结果,有人提出,B细胞记忆出现在多个层级,并具有不同的功能。
Ti B细胞反应基于抗原类型的区别可分为两种Ti-1和Ti-2。Ti-1抗原,例如细菌脂多糖(LPS),可以不考虑抗原的特异性而直接诱导B细胞的激活,它们还通过Toll样受体为B细胞提供第二个信号。Ti-2抗原,例如,肺炎球菌多糖或抗原2,4-二硝基苯偶联右旋糖酐(DNP-DE),都是高度重复的结构,可以交叉连接足够数量的BCR以完全激活抗原特异性B细胞。Ti-1抗原可以激活未成熟和成熟B细胞,而Ti-2抗原只激活成熟B细胞。Ti-2 B细胞反应主要由B1和MZ B细胞执行,并定位在滤泡外病灶。
多年来,人们认为针对Ti抗原的免疫反应不可能产生免疫记忆。早期的研究表明,初次免疫后,再次接触DNP-DE,产生的抗DNP抗体效果很差。然而,这种无反应性并不是由于缺乏抗原特异性的记忆B细胞,而是因为产生了抑制B细胞激活的半抗原特异性抗体。为了证明这一点,将DNP-DE诱导的脾脏细胞过继转移到已辐照的受体上,然后再进行刺激,结果是IgM反应增强。最近,有研究表明,B1b细胞在对Ti抗原的反应中会产生记忆B细胞,另外,B1a细胞似乎也会产生类似记忆细胞的特征。与Td B记忆细胞相比,Ti记忆B细胞在某些标志物方面出现了不同的表型。
小鼠模型中存在自身抗体会产生致病性,导致一些免疫性疾病,如系统性红斑狼疮(SLE),I型糖尿病和类风湿性关节炎(RA)。然而,产生的自身抗体本身并不一定会诱发自身免疫性疾病,相反,这些疾病的复杂的病理表现是由多个基因组合控制的。这些模型中的抗体可能来自于自体反应的记忆B细胞,尽管由于缺乏可靠的表面标记,这些细胞很难被定义和跟踪,因此大多数研究都依赖于‘传统的’记忆B细胞表型以及杂交瘤的建立。有证据表明,记忆B细胞和自身抗体都是在次级淋巴器官、受影响的局部器官中、GCs中以及在滤泡外的聚集物中形成的。
自身免疫性风湿病最常用的标志物之一是类风湿因子(RF),一种针对IgG的Fc部分的自身抗体。例如存在于易患狼疮的MRL/lpr小鼠中的自身抗体。这些自身抗体经历了CSR和SHM,而且RF特异性B细胞反应与外源性抗原引起的记忆B细胞反应非常相似。通过一个模型系统显示,具有针对RF(AM 14)特异性BCR的TG B细胞是以T 细胞依赖的方式被激活的,这发生在脾脏的T细胞区和红髓的边界,而不是在GCs中。与上面讨论的Td记忆B细胞相似,自身反应性的AM 14 B细胞可以进一步发展为CD73阳性的记忆B细胞,也可以发展为生命周期短的浆细胞。
B细胞在GCs中的存活取决于各种因素,包括细胞死亡受体Fas。该受体可以消除GC中的非特异性和自发性的B细胞;因此,如果Fas或FasL信号通路被破坏,自体反应记忆B细胞和浆细胞的生存和产生就会被允许。相反,在选择抗原特异性非自体反应的B细胞的过程中,其他逃逸信号确保了对Fas介导的细胞凋亡的抵抗。事实上,MRL/lpr小鼠出现的类似系统性红斑狼疮的综合征,包括肾小球肾炎、多动脉炎、关节炎和咽喉炎,是由于缺陷的Fas基因(lpr)与其它身份不明的突变基因的结合。
已知GCs的自发形成发生在自身免疫性小鼠的次级淋巴器官中,如自发性糖尿病NOD小鼠和狼疮模型 (MRL/lpr, PN, NZB, NZB/W, B6/lpr and BXSB 雄性小鼠),在没有免疫或感染的情况下,这些1至2个月年龄的小鼠已经发生了。基于用抗CD40配体抗体治疗后GCs的消退,在自身免疫中形成的GCs和由免疫引起的GCs都是T细胞依赖性的。已知在自身免疫环境中GCs的自发形成也发生在受影响的器官中,例如在糖尿病NOD小鼠的胰岛和胶原蛋白诱导的关节炎(RA最常用的小鼠模型)的滑膜组织中。在糖尿病NOD小鼠中,胰岛的GCs与次级淋巴器官中的GCs非常相似,因为它们都含有FDCs和T细胞,存在B细胞上调AID以及表达体细胞突变的寡克隆BCR组合。此外,GC B细胞可以在原地分化为可以产生针对胰岛素的抗体的浆细胞,也很可能分化为记忆B细胞。在患有胶原蛋白引起的关节炎的小鼠的关节中,大约50%的B细胞表达了GC标志物,如GL7和Fas,而同一小鼠脾脏中GC B细胞的相应比例约为10%。因此,很可能小鼠自身的GCs有助于记忆B细胞和浆细胞的产生。
综上,用于研究小鼠记忆B细胞的模型系统对实验结果很重要,因为它们在初次(和再次)免疫反应的持续时间、GCs的持久性和记忆亚群方面存在差异。实验结果还取决于抗原的剂量和类型、免疫的时间间隔以及用于定义记忆B细胞的标记物。然而,之前的数据表明,记忆B细胞的形成有两种途径,一种是是GC依赖性的,另一种不依赖。这两种途径都需要T细胞的帮助,并产生IgM和同型转换的记忆B细胞。如所讨论的转换和非转换记忆B细胞的五个亚群,也可能符合这两种途径,也许代表免疫反应的不同阶段。沿着其中一条途径,记忆B细胞将被生成并表达未变异的抗体,保护宿主免受各种入侵者变异体的伤害,而另一种途径将产生记忆性B细胞,它能以高亲和力、变异和同型转换的抗体迅速作出反应,并提供对同一抗原的再次变异的防御。Ti抗原也可以在同型转换和非转换B细胞中产生记忆反应。
在自身免疫条件下,自体免疫反应最初可能与那些外源性抗原所驱动的途径一样。然而,由于致病的自身抗体主要是突变的和同型转换的,这可能表明自身抗原的持续存在使反应偏向于长期的GCs和永久产生GC依赖性记忆B细胞和自身抗体产生的浆细胞。
决定B细胞命运的机制,也就是说,是什么使细胞走早期记忆B细胞而不是GC B细胞的途径,以及什么使GC B细胞分化为记忆B细胞而不是浆细胞,目前仍不清楚。到底是一个信号,还是几个信号将B细胞引向某条道路,目前还不完全清楚,也许是在内在和外部信号的影响下,例如抗体反馈机制。或者,决定性的事件可能只是随机的,例如,细胞分裂时抗原的不平等分配,产生的后代具有不同的呈现抗原和竞争T辅助细胞的能力。
参考文献
Bergmann B, Grimsholm O, Thorarinsdottir K, et al. Memory B Cells in Mouse Models[J]. Scandinavian Journal of Immunology, 2013, 78(2).
胶体金是由氯金酸(HAuCl4)在还原剂作用下形成的纳米级金颗粒(直径1~150nm)分散体系,其颜色随粒径变化呈现橙黄色至紫红色。胶体金因具有高电子密度、稳定性及与生物大分子(如抗体、抗原)的静电结合能力,被广泛应用于免疫检测、生物标记及诊断试剂开发。
制备颗粒均匀、分散度好的胶体金非常关键,胶体金颗粒直径的变异范围太大会影响试验的稳定性和重复性。如胶体金颗粒的形状不规则或粒径不均一,胶体金标记物容易解离、沉淀,从而产生金标扩散不完全、反应区底色过深和假阳性现象。胶体金的制备过程中的细节关乎制备的成败。首先是操作环境和所用容器的清洁度。操作环境应保持清洁无尘粒,最好有专用工作区。所用玻璃容器必须绝对清洁,玻璃表面少量的污染会干扰胶体金颗粒的生成,产生凝集颗粒,玻璃器皿用前要经过酸处理、超声洗涤处理,并用蒸馏水、超纯水依次冲洗浸泡,然后烘干备用。
胶体金的制备一般采用还原法,其原理是利用还原剂将氯金酸溶液中的金离子还原成金原子。常用的还原剂有柠檬酸钠、鞣酸、抗坏血酸、白磷、硼氢化钠等,根据还原剂类型以及还原作用的强弱,可以制备5~150nm不等的胶体金。一般还原剂用量越大制备的胶体金颗粒越小。如制备颗粒直径在5~12nm的胶体金溶液用白磷或抗坏血酸,制备大于12nm直径的胶体金则用柠檬酸钠。
| 粒径(nm) | 0.01%氯金酸(mL) | 1%柠檬酸三钠(mL) |
|---|---|---|
| 16 | 100 | 2.00 |
| 24.5 | 100 | 1.50 |
| 41 | 100 | 1.00 |
| 71.5 | 100 | 0.70 |
由于胶体金颗粒在电解质中不稳定,制备后应立即用大分子(如蛋白质)进行标记。胶体金颗粒对蛋白质的吸附作用取决于pH值。在pH值=pI值时,蛋白质溶解度最小,水化程度最小,最容易吸附到疏水的金颗粒表面,但在实际操作中,一般胶体金溶液的pH值调到稍高于标记用蛋白质的PI值,这样蛋白质带正电,结合更稳定,可用0.1mol/L K2CO3或0.1mol/L HCL调节胶体金溶液的pH至选定值,但通常最适反应pH往往需经多次试验才能确定。在调节胶体金的pH值时,胶体金会阻塞pH计的电极,因此可用普通PH试纸先调到目标pH值附近再换用精密pH试纸调节,也可以用胶体金专业pH计调节pH。标记应在磁力搅拌下,逐滴加入待标记蛋白溶液,混匀30min后继续在磁力搅拌下加入10%BSA,使其终浓度为0.5%,搅拌混匀15min,再加入5%PEG 20000至终浓度为0.1%,再持续搅拌混匀15min,最后4℃静置过夜。
由于盐类成分能影响胶体金对蛋白质的吸附,并可使胶体金聚沉,因此待标记蛋白质溶液若含有较高的离子浓度,应在标记前先对低离子强度的双蒸水透析去盐,并通过离心及微孔滤膜过滤以除去细小颗粒,然后通过系列稀释法找出能使胶体金稳定的待标记蛋白的最低浓度,这一浓度再加10%即为最佳标记蛋白量。标记好的胶体金还应加入终浓度为0.05%~0.1%的PEG 6000或PEG 20000作为稳定剂。
多种蛋白质、葡聚糖、PEG20000、明胶等均为良好的高分子稳定剂,PEG和BSA是最常用的稳定剂。稳定剂有两大作用,一为保护胶体金的稳定性,使之便于长期保存;二为防止或减少免疫金复合物的非特异性吸附反应。稳定剂的合理选择十分重要,不适当的稳定剂有时也会导致非特异性反应。
标记好的胶体金中往往还含有未结合的蛋白质、未充分标记的胶体金以及标记过程终形成的各种聚合物。因此标记好的胶体金还需经过纯化才能使用,一般纯化方法有离心法和凝胶过滤法等。
采用离心法时,一般颗粒10nm以上的胶体金可高速离心,颗粒小于10nm的胶体金要用超速离心,在4℃离心15min至1h不等,弃上清,沉淀用原体积的0.02mol/L TBS pH8.2(含1%BSA,0.05%叠氮钠)溶解,重复离心2~3次,沉淀溶于原体积的1/10 PBS溶液中,4℃保存备用。
凝胶过滤法为纯化免疫胶体金的最好方法,过滤的胶体金颗粒比较均匀,不容易凝集,而离心法转速高,时间长,胶体金颗粒沉淀后容易凝集,用凝胶过滤法克服了这一弱点。将浓缩好的免疫胶体金先以1500r/min离心除去大的聚合物,取上清液过柱。可用Sephacryls-400或Sepharose-4B(或6B)装柱,0.02mol/L TBS pH8.2平衡和洗脱。
免疫胶体金技术(Immune colloidal gold technique, ICG)是一种常见的标记技术,它是以胶体金为标记物,利用特异性抗原抗体反应,在光镜电镜下对抗原或抗体物质进行定位、定性乃至定量研究的标记技术,是继三大标记技术(荧光素、放射性同位素和酶)后发展起来的固相标记免疫测定技术。免疫胶体金技术作为一种新的免疫学方法,因其快速简便、特异敏感、稳定性强、不需要特殊设备和试剂、结果判断直观等优点已在体外诊断、食品安全、环境监测等领域取得广泛的应用,如妊娠试验、传染病病原抗体的检测、蛋白检测和药物测定等,其在兽医临床上的应用也日益广泛。
氯金酸(HAuCl4)在还原剂作用下,可聚合成一定大小的金颗粒,形成带负电的疏水胶溶液,由于静电作用而成为稳定的胶体体系,故称胶体金。胶体金颗粒散在,直径从几纳米到几十纳米不等,由于不同直径的胶体金的光散射各异,所以其溶胶颜色的深浅相应发生显著的变化。
胶体金标记实质上是蛋白质等大分子被吸附到胶体金颗粒表面的包被过程,吸附机理可能是胶体金颗粒表面负电荷,与蛋白质的正电荷基团因静电吸附而形成牢固结合。用还原法可以方便地从氯金酸制备各种不同粒径、也就是不同颜色的胶体金颗粒,这种球形的粒子对蛋白质有很强的吸附功能,可以与葡萄球菌A蛋白、免疫球蛋白、毒素、糖蛋白、酶、抗生素和激素等多种物质非共价结合,从而使其成为免疫反应的优良标记物。
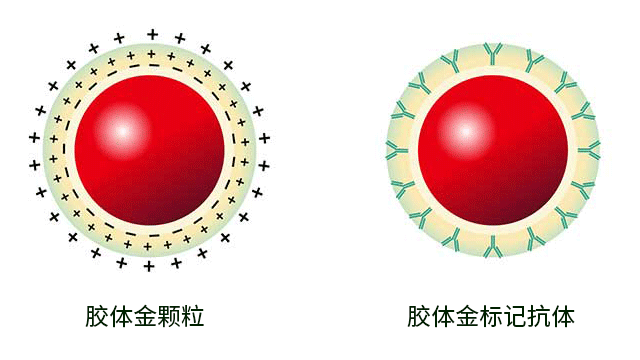
图1 胶体金结构图
免疫胶体金颗粒具有高电子高密度的特性,在显微镜下金标蛋白结合处,可见黑褐色颗粒,当这些标记物在相应的配体处大量聚集时,肉眼可见红色或粉红色斑点,因而可用于定性或半定量的快速免疫检测,而且金颗粒还可催化银离子还原成金属银,在胶体金免疫测定时加入银染色液,能放大反应信号,大大增加测定的灵敏度。
近年来,研究人员根据胶体金的物化性质进一步拓展了基于胶体金标记的生物检测技术及胶体金在其他生物学方面的应用。目前已广泛应用于被动凝集试验、光镜染色、免疫印迹、流式细胞术、液相免疫测定、斑点金免疫渗滤及免疫层析等,其中在医学检验中的应用主要是胶体金免疫层析法(immune chromate graphic assay, ICA)和斑点免疫金渗滤法(Dot immune gold filtration assay, DIGFA)。
胶体金免疫层析技术近年来发展迅速,在生物医学领域特别是医学检验中得到了广泛应用。胶体金免疫层析试纸条主要是将特异性的抗原或抗体以条带状包被在NC膜的检测T线上,胶体金标记的抗原或抗体吸附在结合垫上,当待测样品加到试纸条一端的加样孔上后,通过毛细作用向前移动,溶解结合垫上的胶体金标记的抗原或抗体,再移动至包被的抗原或抗体的检测线处,如果样品中含有相应的抗体或抗原,包被在检测线上的抗原或抗体和胶体金标记物与样品中的相应抗体或抗原结合,形成免疫复合物,胶体金富集在检测线处形成一条可见的紫红色线。如果待检血清中没有相应抗体,胶体金标记物将不会与包被在检测线上的抗原或抗体结合,胶体金不会富集,检测线上不会出现紫红色色线。当样品与胶体金标记物继续往上移动至质控线时,就与包被在质控线处的特异性抗体或抗原结合,在质控线上形成免疫复合物,出现一条胶体金富集的紫红色色线。
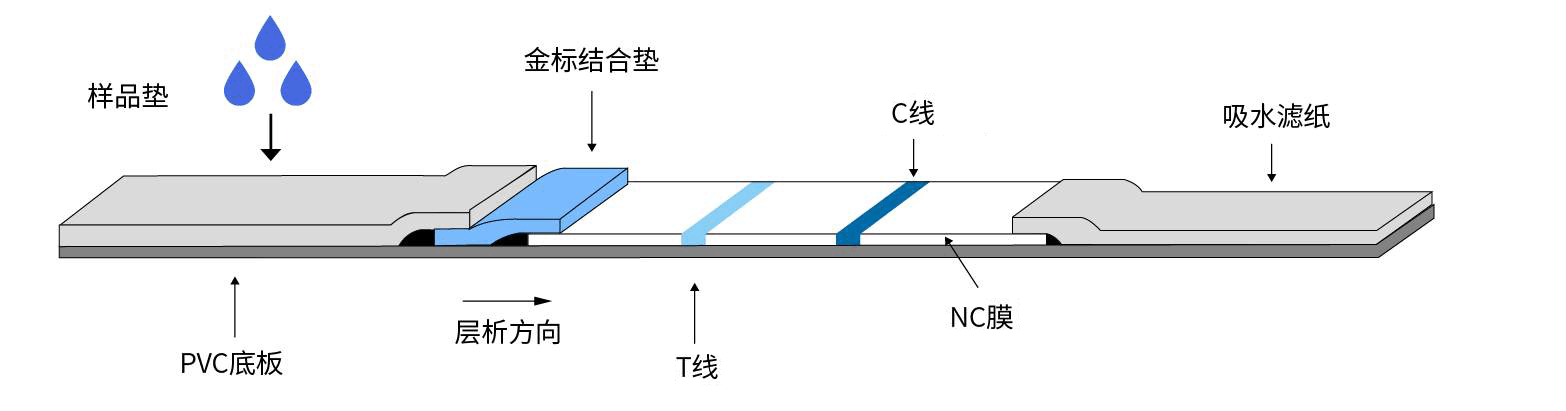
图2 胶体金免疫层析试纸条结构图
这种检测技术具有操作简单快速,特异性、敏感性好,可单份测定,结果直观,无须特殊仪器等优点,适合于广大基层单位、医院、野外作业人员以及大批量时间紧的检测和大面积普查等,具有巨大的发展潜力和应用前景。
常用的胶体金免疫层析检测方法有双抗夹心法、竞争法、间接法。
此方法常用于检测生物大分子和颗粒抗原。需要制备分析物的配对抗体,包被抗体用胶体金标记,固定在结合垫上,捕获抗体固定在NC膜的检测线上。此外,还需要制备与金标抗体特异性结合的二抗,固定在NC膜的质控线上。
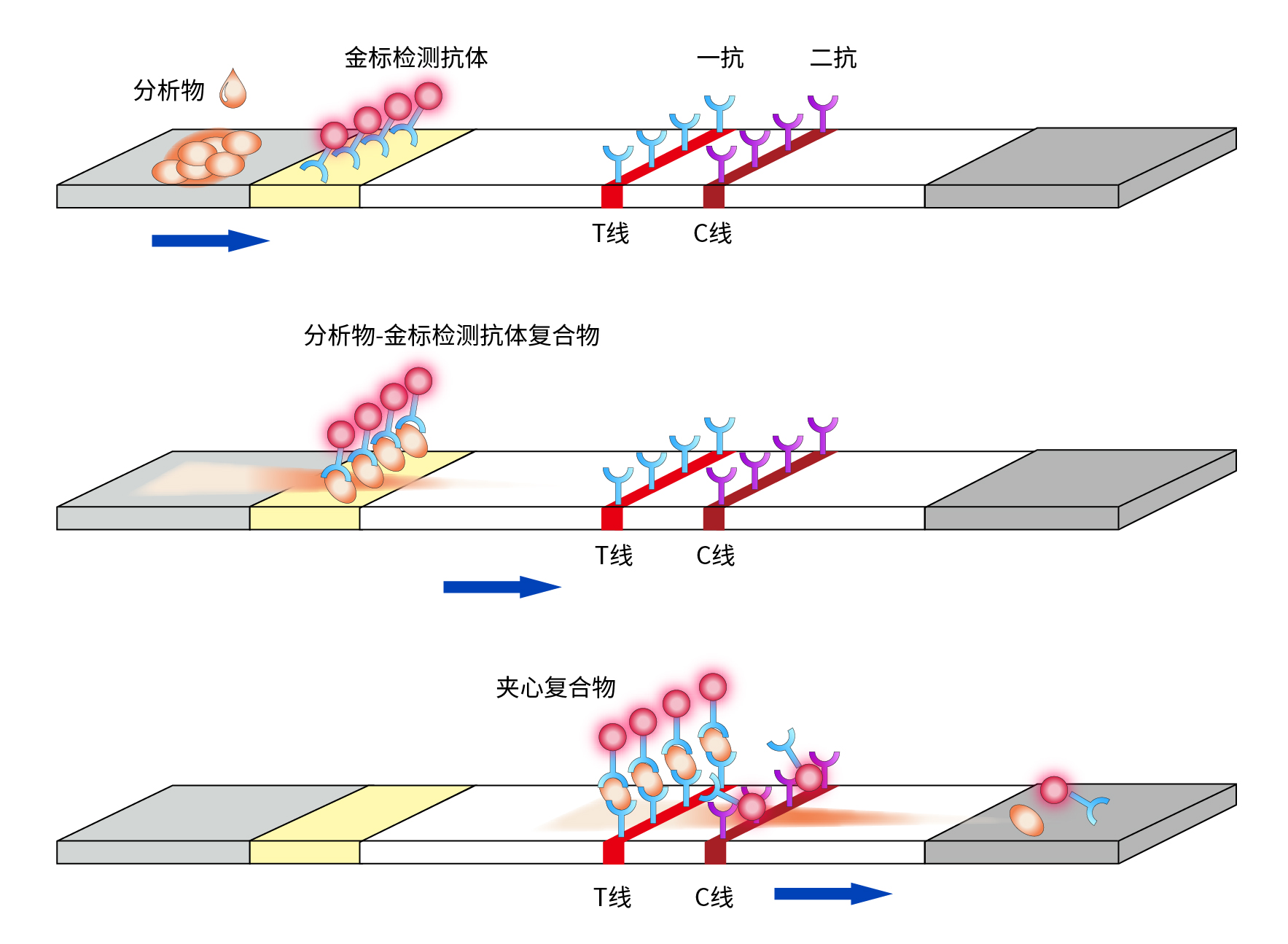
图3 双抗夹心法
此方法常用于小分子抗原的检测。将胶体金标记的检测抗体固定在结合垫上,待测抗原固定在NC膜的检测线上,同时需要将金标抗体特异性结合的二抗固定在质控线上。
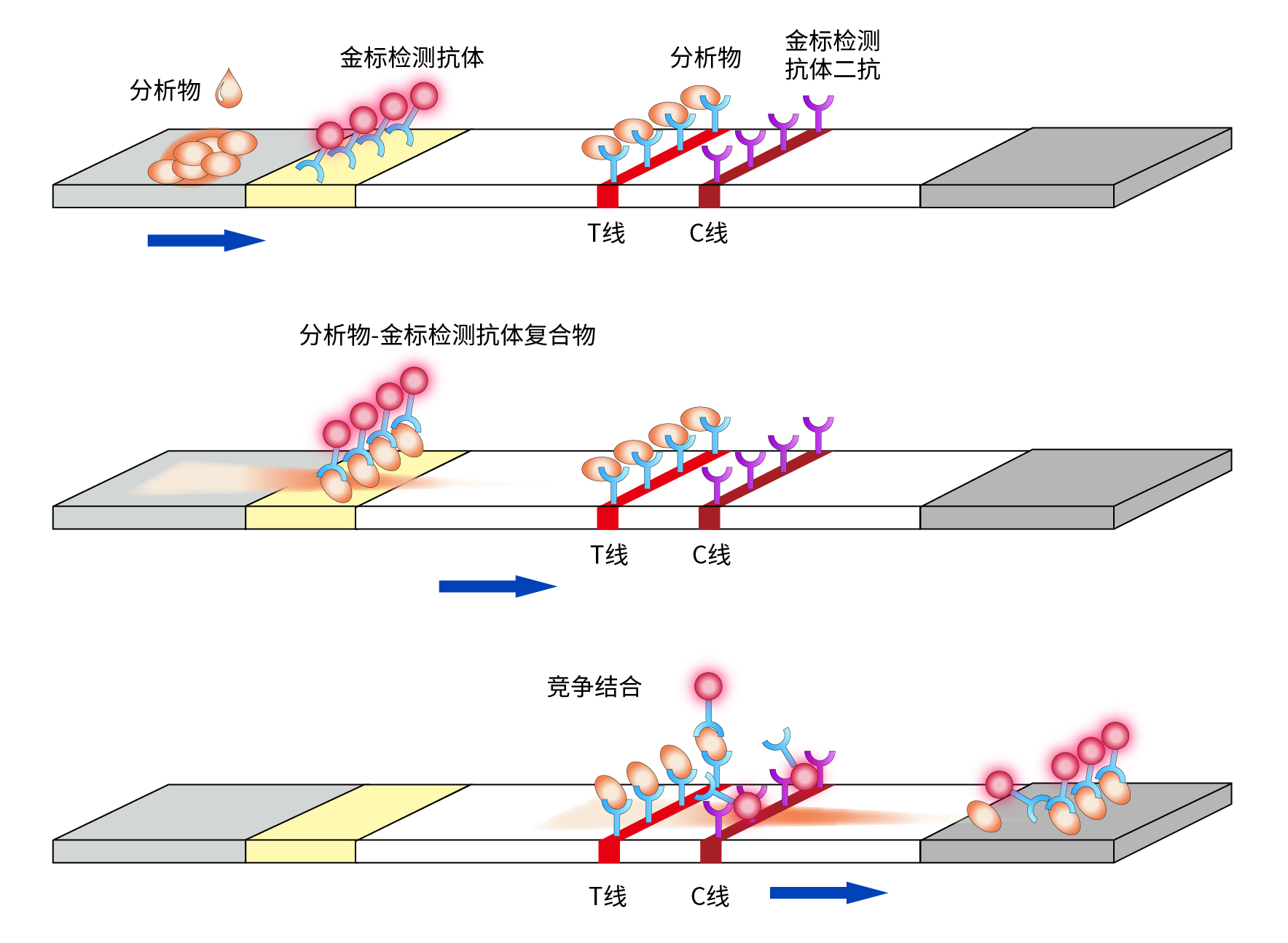
图4 竞争法
此方法主要用于抗体的检测。将能与抗体非特异性结合的Protein A用胶体金标记后固定在结合垫上,与待测抗体特异性结合的抗原固定在NC膜的检测线上,并将抗Protein A的抗体固定在NC膜的质控线上。
斑点免疫渗滤技术是20世纪80年代中期从固相酶免疫测定技术发展起来的较简便的固相标记免疫测定方法,该技术主要应用NC膜作载体的免疫检测技术,先将抗原或抗体点于NC膜上,封闭后加待测样品,洗涤后用胶体金探针检测相应的抗原或抗体。通过金颗粒来放大免疫反应系统,使反应结果在固相载体NC膜上显示出来。与间接过氧化物酶法相比,免疫胶体金斑点渗滤法要更为敏感。
胶体金制备容易,价格低廉。免疫金标记不仅可用于常规光镜,而且还可用于荧光显微镜,运用光镜的IGSS法是迄今最敏感的免疫组化方法。由于金颗粒具有很强的激发电子能力,还可用于扫描电镜和X射线衍射分析等。胶体金可以标记多种生物大分子物质,如抗体葡萄球菌蛋白A(SPA)、凝集素、多糖、多肽及其它蛋白质而不影响其生物活性。免疫胶体金对组织细胞的非特异性吸附作用小,故具有较高的特异性。胶体金是高电子密度颗粒性标记物,电镜下分辨率高,对超微结构遮盖少,并易与其他颗粒性结构相区别,具有较精确的定位能力。金颗粒大小可以控制,颗粒均匀,可进行双重和多重标记,即用不同大小的金颗粒分别标记不同的抗体,实现在同一张切片上观察两种以上的抗原,也可以和其他标记物配合进行双重或多重标记。由于胶体金本身有鲜艳的橘红色,可用光镜或肉眼观察试验结果,也可用分光光度计测定光吸收,进行定量分析。可在切片不同视野中根据金颗粒的数目来半定量抗原。应用于快速检测技术还具有操作简单快速,特异性、敏感性好,可单份测定,结果直观,且可保存试验结果,无须特殊仪器等优点。
1960年Yalow和Berson首次描述了一种使用与放射性信号相连的抗体介导的检测技术,即放射免疫测定技术(radioimmunoassay,RIA)。然而,由于健康风险不得不寻求不涉及放射性信号的方法。某些酶-底物组合产生可量化的颜色变化的发现促进了免疫检测的转变,研究者开发了与抗体连接的酶-底物组合,可以用于检测特定分析物。1971年,欧洲的两个独立研究小组发表了论文,分别描述了进行酶联免疫吸附测定(ELISA)的分步实验过程。
ELISA方法用于检测和定量样品中的特定物质,通常是抗原。将抗原直接或通过称为“捕获抗体”的特异性抗体固定在微孔板中。添加一抗,形成抗原-抗体复合物。一抗或直接用酶标记(即直接ELISA),或通过与酶标记二抗结合(即间接ELISA)。在每个步骤之间,用缓冲液洗涤孔。底物的加入产生指示样品中抗原存在的颜色信号。光密度的测量与样品中抗原的量成正比。
直接ELISA代表用于检测抗原的最简单的一种方法。将含有待测分析物溶液加入到96孔板中,使其在包被在孔内(图1a)。通常使用碳酸盐-碳酸氢盐包被缓冲液,有助于抗原被动吸附到孔。缓冲液pH维持在至少为9,使抗原保持可溶,并确保它们具有可与带正电的板结合的总负电荷。
同时进行标准品、阳性对照和阴性对照的准备。标准品是通常由ELISA试剂盒提供的含有已知浓度分析物的样品。通过在接近预期未知浓度的范围内连续稀释已知浓度的分析物来制备标准曲线。当对血清样品进行ELISA时,通常建议在血清中稀释第二标准曲线,以评估血清中的其他蛋白质是否影响检测(即加标对照)。阳性对照为含有分析物的可溶性样品或纯化蛋白。阴性对照为已知不表达检测到的分析物的样品。
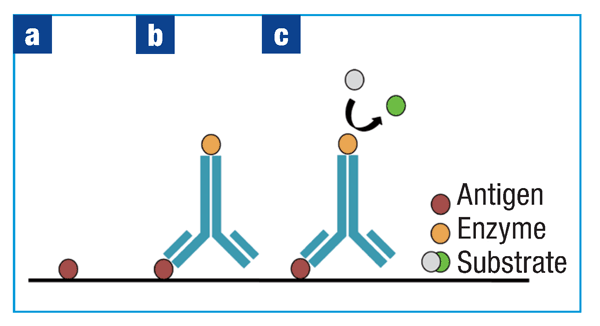
图1 直接ELISA
在抗原包板后,可以处理掉多余的溶液,并加入封闭蛋白覆盖未被抗原包被的孔表面,以限制非特异性结合。在步骤与步骤之间,按照试剂盒的指示,用中性pH的磷酸盐缓冲液洗涤孔,去除未结合的测定组分,这使检测过程中的背景最小化,并提高了测定的特异性。
封闭缓冲液对于饱和未被占用的结合位点是必要的,从而可以最大限度地减少非特异性结合和非特异性蛋白质-蛋白质相互作用。尚未确定标准化封闭缓冲液是否适用于所有试验。理想的封闭缓冲液必须与其他测定组分无交叉反应性,最大限度地减少变性并表现出低酶活性。封闭缓冲液主要包括两类:蛋白质(例如牛血清白蛋白、酪蛋白)和去垢剂(例如吐温20、Triton X-100),需要进行测试以选择最佳的封闭缓冲液并优化流程。封闭缓冲液的选择主要受特定分析成分和孔板表面化学性质的影响。
在此步骤之后,直接和间接ELISA在方案上存在差异。在直接ELISA中,加入偶联酶的检测抗体,使其与包被孔的抗原结合(图1b):将孔板孵育足够长的时间允许抗原-抗体结合,并用磷酸盐缓冲液洗涤除去多余的抗体。然后加入酶的底物,在黑暗环境中为酶-底物相互作用留出足够时间,并用特定溶液停止反应。酶-底物作用形成的颜色变化可以通过酶标仪检测(图1c),并将样品读数与标准曲线进行比较。
在间接ELISA中,抗原固定、封闭和洗涤后(图2a),加入与抗原结合的一抗(图2b):使用碳酸盐-碳酸氢盐包被缓冲液将一抗附着到板上。然后孵育孔板并用缓冲液洗涤,随后加入阻断剂和偶联酶的二抗(图2c)。孵育孔板,洗涤,加入底物,并使用酶标仪扫描孔(图2d)。也可以用荧光标记二抗,并使用荧光计在紫外光下对结果进行定量,即荧光免疫吸附测定(FLISA)。
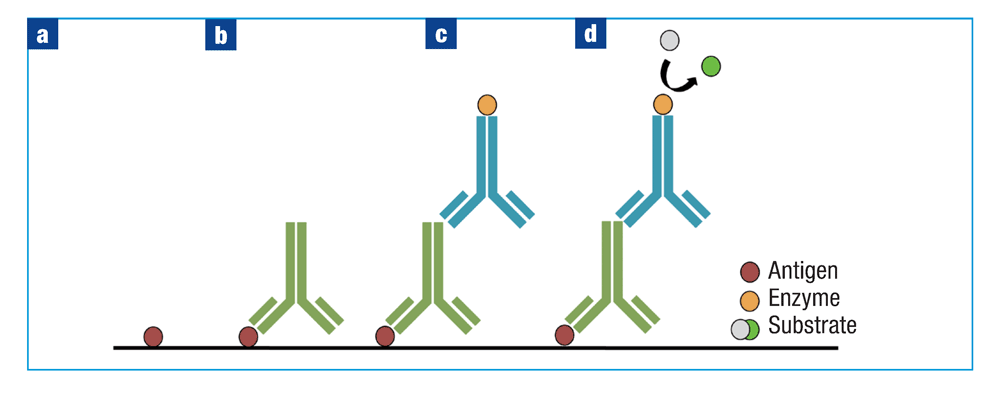
图2 间接ELISA
直接ELISA是一种快速诊断工具。然而,它不能信号放大,因此灵敏度不高。当样本中有高比例的抗原需要评估时,可以优先使用这种方法。抗生物素蛋白/链霉抗生物素蛋白-生物素复合物(ABC)的发展允许分析具有低抗原比例的样品。与荧光团连接的生物素标签用于在间接ELISA中标记第二抗体。亲和素/链酶亲和素-生物素复合物的发展使低抗原比例的样品分析成为可能。每个蛋白分子能够结合多达四个生物素标签,允许多个ABC连接到二抗上,这样可以检测到更强的信号,增加了测定的灵敏度。然而,如果ABC体积太大,可能无法穿透某些组织。
直接ELISA的主要缺点是必须使用特异性抗体,在抗体的选择上没有灵活性,并且标记抗体是耗时且昂贵的。间接ELISA可以使用来自同一个物种的不同一抗,这些一抗可以使用相同二抗标记(易通过商业获得)。然而,二抗有发生交叉反应的可能。所产生的非特异性信号可以使用校准曲线来定量,以相对于被检测的分析物来比较测定。
夹心ELISA分析可以解决一些更复杂的情况,实验的名称源于两种抗体将抗原“夹”在之间的设置(图3)。将含有捕获抗体的包被缓冲液加入到孔板中并使其粘附(图3a)。孵育后,洗涤板并加入封闭缓冲液以封闭孔上剩余的结合位点。然后将样品加入到每个孔中并孵育特定的时间长度(图3b)。
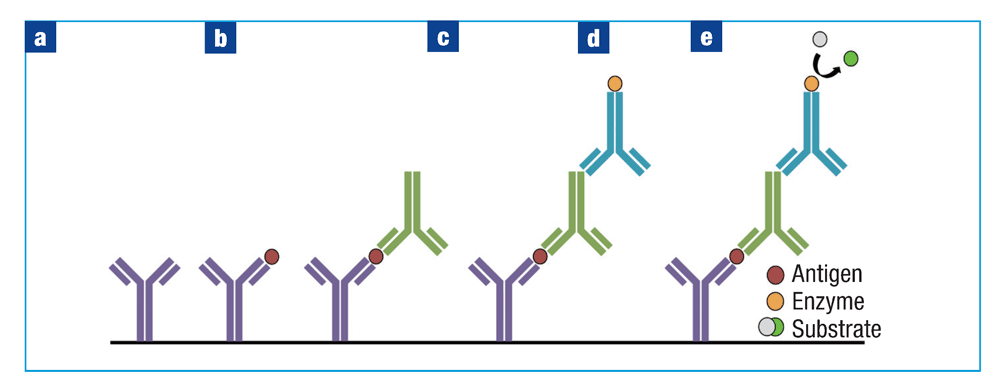
图3 夹心ELISA
为确保结果的准确性,每个孔板必须保留一个标准样品(阳性对照)和一个空白样品(阴性对照)孔。向每个孔中加入检测抗体(图4c),随后加入偶联二抗和封闭缓冲液(图4d)。在每个步骤之间,用磷酸盐缓冲液洗涤孔。加入底物溶液并使用酶标仪检测结果(图4e)。
夹心ELISA的优点是其对分析物检测具有更高的特异性。捕获抗体固定样品中的特异性抗原,因此样品不需要事先纯化,这与直接和间接ELISA不同。通过使用两个抗体“识别”步骤,抗原被有效地夹在捕获抗体和检测抗体之间。夹心ELISA的主要缺点是必须使用特定的捕获抗体和检测抗体。捕获抗体和检测抗体与抗原上的不同表位结合,必须提前进行验证以防止对抗原结合位点的竞争。同时,夹心ELISA也比直接和间接ELISA更耗时。
竞争ELISA不同于直接ELISA、间接ELISA和夹心ELISA,它使用竞争性结合过程。将一抗与未纯化的样品一起孵育(图4a),与样品中存在的抗原结合(图4b)。样本中存在的抗原越多,形成的抗原-抗体复合物就越多。将抗原-抗体复合物加入到预先包被抗原的96孔板中,未结合的抗体与孔中的抗原结合(图4c)。形成的抗原-抗体复合物越多,可用于结合孔中抗原的抗体就越少,即在样品中的抗原和孔中预包被的抗原之间存在抗体的竞争。将孔板孵育一定时间,然后用磷酸盐缓冲液洗涤除去未结合的抗体,并加入封闭缓冲液(图4d)。加入与酶偶联的二抗(图4e)。最后,加入底物并使用酶标仪检测颜色变化(图4f)。
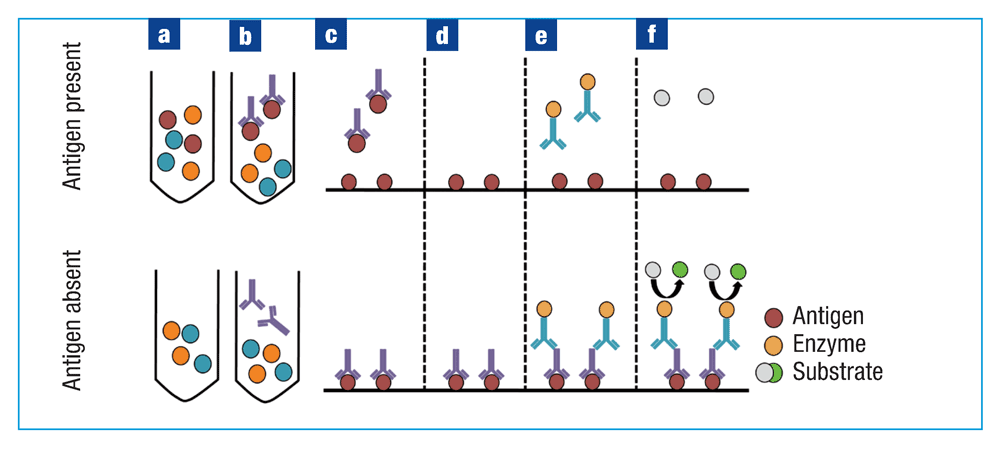
图4 竞争ELISA
竞争ELISA的标准曲线与其他类型ELISA曲线相反,样品中高含量的抗原产生较低的信号。竞争ELISA适用于未纯化的样品,通常只需离心去除微粒即可,并且比夹心ELISA的检测范围更宽。竞争ELISA通常用于检测不具有多个抗原表位的小分子。竞争ELISA的特异性较低,不应用于稀释样品(通常可使用夹心ELISA)。
| 类型 | 优点 | 缺点 |
|---|---|---|
| 直接ELISA |
|
|
| 间接ELISA |
|
|
| 夹心ELISA |
|
|
| 竞争ELISA |
|
|
| 问题 | 可能原因 | 解决方法 |
|---|---|---|
| 低信号(样品和标准曲线) | 使用旧孔板或久置试剂 | 准备新孔板/新配试剂(检查pH值),并适当储存 |
| 洗板过猛 | 用轻轻吸取缓冲液洗板 | |
| 孔变干 | 使用带有密封膜的孔板,或使用封口膜覆盖 | |
| 酶反应不充分 | 优化显影温度和时间 | |
| 检测波长不正确 | 检查酶标仪上的过滤器和软件 | |
| 低信号(仅样本) | 低于检测下线 | 降低稀释系数或浓缩样品 |
| 不兼容的样本类型 | 使用阳性对照确保兼容性 | |
| 低/较差信号(仅标准曲线) | 标准品未正确复溶或储存 | 重新配制新标准品,储存于-70ºC |
| 标准品添加错误 | 检查是否存在移液错误 | |
| 标准品稀释/配制错误 | 检查标准品的配制 | |
| 阴性对照的阳性结果 | 样品污染 | 使用新配试剂/正确使用移液器 |
| 洗涤不充分 | 增加洗涤步骤 | |
| 检测抗体与捕获抗体相互作用(夹心ELISA) | 确保抗体不发生交叉反应 | |
| 高背景 | 背景孔被污染 | 小心移液,使用多通道移液器 |
| 洗涤不充分 | 增加洗涤步骤 | |
| 高信号 | 样本含有高于测定范围的抗原 | 稀释样品 |
| 洗涤不充分 | 增加洗涤步骤 | |
| 过饱和样品 | 减少孵育时间或温度 | |
| 变异系数(CV)高 | 孔板中存在气泡 | 读数前确保无气泡 |
| 移液量不一致 | 使用校准的移液器,小心移液 | |
| 边缘效应(即边缘附近的信号在统计上不同于中心孔) | 确保孔板上各个孔具有相同的温度和湿度 | |
| 样本不均匀 | 移液前彻底混合样品 | |
| 堆叠孔板 | 切勿堆叠孔板 |
生物制品特别是治疗性抗体药物的免疫原性是临床前和临床研究中需要解决的重要问题,它可能导致疗效损失、药物暴露变化甚至严重副作用,且使得毒性、药代动力学(PK)和药效学(PD)数据的解释复杂化。一般通过检测和定量抗药抗体(ADA)来评价生物制品的免疫原性,检测方法主要有直接法和桥连法。直接法中使用药物捕获ADA后加入种属特异性的抗体进行检测,直接法的限制在于存在种属特异性,且无法检测所有同种型的ADA。桥连法中使用药物捕获ADA后加入标记的药物,对形成的药物-ADA-药物复合物进行检测。桥连法不受种属限制,可检测所有同种型的ADA(如IgM和IgG),及除IgG4外大部分亚型而应用更广。但是,桥连法检测ADA易受多种因素影响,血液循环中可溶性靶点是常见的干扰因素之一。可溶性靶点会引起假阳性或假阴性结果,进而影响药物免疫原性风险的评估,因此开发可降低可溶性靶点干扰的ADA检测方法非常关键。
由于疾病导致的生理差异,在血液中可能存在从细胞膜表面脱落的靶点受体、因细胞裂解而释放的胞内靶点等可溶性靶点。可溶性靶点产生影响主要包括:(1)给药前体内高浓度的可溶性靶点可能干扰ADA检测、与真实药前数据偏离及对药物免疫原性风险的判断;(2)个体间可溶性靶点浓度不同,检测结果药前ADA个体差异大,不利于临界值的确定;(3)在某些情况下,虽给药前较低的可溶性靶点浓度不会影响ADA检测,但给药后靶点受体从细胞膜脱离水平增加、内在负反馈机制导致可溶性靶点分泌增多、靶点分子与药物结合导致清除减慢和浓度增加等原因亦可能干扰ADA检测。
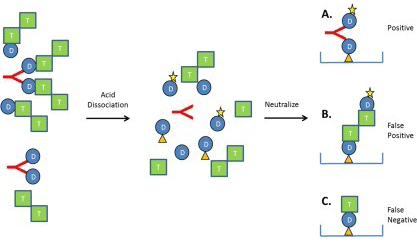
可溶性靶点对ADA检测的影响
图中“D”表示抗体药物、“T”表示药物靶点和红色“Y”形符号表示ADA。酸解离后,药物-靶点和药物-ADA复合物被破坏。(a)在没有干扰的情况下,捕获药物(橙色三角形&“D”)、ADA和标记的药物之间形成桥连。(b)如果药物靶点是多聚体(如双“T”分子),则在没有ADA的情况下也可能会形成桥连,从而导致假阳性结果。(c)如果药物靶点是单体,并且在干扰ADA结合的结构域以高亲和力结合捕获药物,则药物靶点可导致假阴性检测信号。
不同试验条件下,可溶性靶点的物理化学特征及其与药物的相互作用、亲和力大小决定了对ADA检测干扰程度。基质中可溶性靶点形成的二聚体或多聚体直接与捕获试剂和检测试剂桥连而出现假阳性信号;而单体靶点分子与ADA竞争结合捕获试剂导致假阴性结果。此外,采用酸解离“药物-ADA”复合物的同时会释放与药物(如单抗、融合蛋白)结合的靶点分子,造成基质中的靶点浓度迅速升高(可达数量级改变,如机体接受靶向血管内皮生长因子的单抗或融合蛋白治疗时),反而可能会导致异常的假阳性或假阴性结果。
目前,常用于降低可溶性靶点干扰的方法包括多种。在方法开发过程中应仔细考虑并权衡每种策略的优缺点及可能存在的风险,选择易操作、效果显著、风险较小的策略;若单一策略无法满足需求,可尝试将多种策略结合以达到目的。
在筛选实验或确证实验中加入抗可溶性靶点的特异性抗体可降低游离靶分子的干扰。一般情况下,在观察到有可溶性靶点干扰后可首先尝试制备多个特异性抗体,通过实验设计以筛选出可减轻干扰、不影响ADA检测的抗体。该方法简单易操作,通量较高;若在方法学验证期间即采用该方法,可明显降低空白个体的背景值差异,有利于临界值的确定。去除血管生成素、血管内皮生长因子和细胞因子等干扰均可采用该方法。
若采用上述方法,抗可溶性靶点抗体设计需注意其结构序列应与药物骨架不同、或与药物不存在或较少存在同源性序列,否则会造成ADA与抗可溶性靶点抗体的交叉反应。在模拟真实样品情况确定检测方法后,一定要用此方法检测真正样品(临床前或临床),以评估是否会影响ADA检测。
考虑药物的作用机制,若药物通过与靶点结合而抑制靶点与其受体结合,可采用向基质中加入过量的可溶性受体以排除干扰。通常,靶点与其受体亲和力较高,因此加入受体可有效抑制可溶性靶点干扰。有研究表明,ADA检测实验中阳性信号值随加入受体浓度的升高而降低,但不能完全抑制可溶性靶点的影响;而引入弱酸性环境后,低浓度的受体即可完全抑制可溶性靶点的影响。去除抗IgE抗体ADA检测中IgE的干扰也可考虑加入IgE的受体。
这种方法可能出现的问题是,即使加入极其过量的可溶性受体也无法减轻干扰。造成以上情况的原因可能有以下几点:(1)受体的制备技术限制,体外无法生产出和天然受体完全相同的重组受体,导致无法和可溶性靶点有效结合。如天然受体在细胞表面表达,体外生产的重组可溶性胞外区结构可能与其天然结构不同;(2)药物与靶点的亲和力极高,靶点很难与可能低亲和力的可溶性受体结合。除此之外,还应结合受体生产量是否可满足样品检测的需求、受体生产的成本进行考虑采用此方法的必要性。
若不能获得满足要求的针对可溶性靶点的抗体或受体,可尝试将样品中的ADA预先从基质中分离纯化出来,再用于检测。此方法预处理过程可能导致ADA部分丢失,且增加了样品预处理步骤,操作过程较为复杂,试验通量较小。此外,可能需特殊的仪器及耗材。
酸化-磁珠提取(bead extraction and acid dissociation,BEAD)法可较好分离ADA。其主要通过样品进行酸化处理释放所有ADA后加入大量含生物素标记的药物捕获ADA达到目的。BEAD法不仅可有效降低可溶性靶点及基质中其他物质的干扰,也可提高方法的灵敏度和药物耐受程度。但由于多次引入酸化步骤,可能导致ADA降解、结构改变或ADA分离不完全,使ADA检测信号值低于真实值。因此,在方法学开发阶段需确认该法对于ADA检测无影响后,再用于样本检测。
先经过亲和捕获ADA,然后采用桥连接法进行分析,即将样品与加入生物素标记的药物在包被链霉亲和素板上过夜孵育,随后经酸化处理将捕获的ADA解离后进行桥连试验检测也可有效减弱可溶性靶点的干扰。
除了从样品中分离ADA,还可通过固相提取去除可溶性靶点以消除干扰。其基本原理是将样本与包被了特异性结合可溶性靶点的单抗或配体的磁珠或板共孵育后,离心去除磁珠或从板吸出溶液即可去除可溶性靶点。
凝集素可特异性地结合糖基化复合物的糖基,因此加凝集素后能抑制糖基化可溶性靶点造成的ADA检测干扰。、需注意的是,由于凝集素可与人免疫球蛋白的糖基结合,需评估加入凝集素是否会同时干扰真实的ADA检测。
若抗体药物的Fc结构域含Pro329Gly修饰,可利用该修饰位点设计ADA检测方法,避免基质中可溶性靶点和过量药物的干扰。加入过量药物,使基质中全部游离的ADA与药物形成复合物;随后采用生物素标记的抗Pro329Gly的抗体捕获ADA-药物复合物;加地高辛标记的hsFcγRI进行检测。
与常规桥连试验相比,该法可特异性地检测ADA,有效避免可溶性靶点的干扰;对于天然抗体的检测灵敏度较好,有利于评估给药后的ADA产生。但该法需对抗体Fc进行Pro329Gly修饰,仅能检测IgG1抗体,不能检测其余ADA亚型,因此适用范围极小。
可溶性靶点的存在会影响药物ADA的检测及结果判断,进一步影响对药物药动学、有效性及安全性的评估。可溶性靶点对生物制品ADA检测的干扰受多种因素影响,包括药物及靶点的物理化学特点、药物与靶点的相互作用、药物的作用机制、选择的实验方法等。开发可减轻可溶性靶点干扰的分析方法非常重要,也颇具挑战。基于风险评估,在ADA方法开发初期,应根据药物的作用机制、靶点的生物学特点、给药后的生理代偿等提前评估相关可溶性靶点对检测的可能干扰。若可溶性靶点干扰大,应充分参考靶点及药物作用特性,尝试多种方法,以选择最方便经济可靠的方法去除可溶性靶点干扰,同时不影响真实ADA检测。在选择特定方法后,应用真实样本检测以进行进一步佐证。需注意的是相同靶点的药物,可溶性靶点对检测的影响也是有差异的,应经完全评估后才能应用于新药物的ADA检测。
参考文献
[1]GUNN G R, SEALEY D C F, JAMALI F, et al. From the bench to clinical practice: understanding the challenges and uncertainties in immunogenicity testing for biopharmaceuticals[J]. Clinical and Experimental Immunology, 184(2), 2016, 137–146.
[2]SHAO X, LUO WJ, WANG HX, et al. Challenges and strategies for anti-drug antibodies detection by bridging assay of biological products with soluble targets[J]. 33(11), 2019, 1007-1012.
基于杂交瘤细胞的单克隆抗体规模生产主要分为体内方法和体外方法。体内方法通常指的是利用活体动物生产单克隆抗体,最常用的是小鼠腹水法。体外方法指的是在细胞培养瓶或生物反应器中进行杂交瘤细胞的培养,从而生产单克隆抗体。
小鼠腹水法操作简单,通过将杂交瘤细胞接种于小鼠腹腔内,让细胞在腹腔内生长并产生腹水,从而获得大量的单克隆抗体。这种方法获得的抗体中常混有小鼠的各种杂蛋白,多数情况下需要纯化后才能使用,并且存在污染动物病毒的风险。
影响体内生产和优化的变量包括小鼠的年龄、性别、宿主品系、接种杂交瘤细胞数量、腹水采集次数以及引物的类型和体积,可以优化这些变量来调整腹水产量和单克隆抗体浓度。例如,产生少量腹水的亚克隆通常只在腹膜腔中形成几个大肿瘤,而产生大量腹水的亚克隆形成大量小肿瘤集落,在整个肠系膜中广泛生长。因此,应选择形成侵袭性较小的软肿瘤的克隆。从一组小鼠中顺序穿刺可以获得最高的单克隆抗体产量和最高的浓度。除了只允许一次腹水采集的侵袭性很强的细胞系外,顺序穿刺通常会将每克单克隆抗体所需的小鼠数量减少2~3倍。允许穿刺的次数应基于小鼠的临床状况,一般来说,最大值应为三次。
优化体内生产需要减少细胞系的侵袭性,以便所有小鼠在腹水产生后能继续存活。常用的两种方法包括选择合适的克隆或改变注射到小鼠腹膜腔中的杂交瘤细胞浓度。产生的单克隆抗体的体积和浓度取决于所选的克隆,因此获得最大体内产量的最佳方法是在小鼠中筛选并使用最佳产量的克隆。体内细胞生长条件是最佳的,几乎所有细胞系都会产生抗体,即使它们没有经过优化。这就是为什么注射到小鼠中通常会挽救难以在体外生长的细胞系。
杂交瘤细胞属于半贴壁性质的细胞,既可以进行单层细胞培养,也可以进行悬浮培养。体外极小规模生产(小于10g)通常使用简单的低密度细胞培养系统,通过将杂交瘤细胞加入培养瓶中,以含10~15%小牛血清的培养基培养,收集培养上清,上清中单克隆抗体含量约10~50 μg/mL。然而,这种方法生产的抗体量有限,大量生产需要进行杂交瘤细胞的高密度培养。对于小规模和中规模生产(10~200g),通常使用中空纤维系统、细胞悬浮培养瓶或滚瓶培养,进行大规模生产(超过100g),可以选择在搅拌式生物反应器、灌注式生物反应器、气升式生物反应器或连续培养系统中进行均质悬浮培养。
体外生产不使用活体动物,同时可以通过细胞驯化使用低血清或无血清培养基进行生产,减少了外来抗原(但使细胞系适应低血清或无血清培养基,通常对细胞系有轻微的抑制作用)。随着一次性材料成本的进一步降低和技术变革提高生产效率和降低设备成本,体外生产的成本应进一步降低。
体外方法失败的最常见原因之一是难以保证细胞培养过程中操作的一致性,例如培养器皿、设备、培养基的灭菌以及系统中的湿度和温度控制。在中大规模中,必须进行严格的程序和环境控制,以最大限度地减少系统微生物污染造成的损失,这些可能是增加体外单克隆抗体生产成本的原因。
单克隆抗体生产三大商业用途分别为诊断、治疗以及新型治疗药物研究和开发,所需的单克隆抗体量以及成本、时间和合规性等因素的考虑取决于目的。商业利益认为0.1~10g的抗体生产规模为小规模,10~100g为中等规模,超过100g为大规模。竞争非常激烈的诊断行业关注成本、周转时间和监管要求,规模通常为中小型,很少达到大型规模。与诊断行业相比,治疗行业对成本和周转时间的关注要少得多,其生产规模为中大型。由于受到高度监管,对任何程序变更都非常敏感。开发治疗药物的biotech单克隆抗体生产规模为中小型,他们比治疗行业更关心生产成本,但关心程度不及诊断行业,同时更关注周转时间,这是因为产品的生命周期较短,需要获得率先进入市场的机会。
商业单克隆抗体生产需要的不仅仅是大批量细胞的培养或将其注射到大量小鼠中,还需要大量的生产前工作,以确保细胞系稳定,能够生产出适当数量的稳定抗体。还涉及搭建用于体内和体外生产以及抗体加工的高质量设备平台,设立质量控制和质量保证部门来满足商业产品所需的良好生产规范的要求。产品批次测试对于确保产品可重复性是必要的。生产过程验证和记录对于保护消费者同样如此。FDA在其法规《人用单克隆抗体产品制造和测试中的考虑要点》(FDA1997)中要求进行生产过程验证和记录。
考虑到商业可行性,体内生产通常更具有经济性。然而,随着单克隆抗体量的增加,现有的体外生产技术可能更加经济,因为与体外生产相关的一些较高固定优化成本(选择具有最佳生长和生产特性的亚克隆以及在低血清或无血清条件下生长相关的成本)被分摊到更大的生产量上,使每克成本与体内生产相比具有竞争力。在大规模生产运行中,如果可以使用无血清培养基,它们可以减少动物使用并减少污染性外来抗原的存在。当单克隆抗体生产的时间很关键且需要量少时,选择体内生产,因为它只需要6周。对于体外系统,时间需求则差异很大。商业数量的单克隆抗体体外生产比体内生产需要更多的时间,因为工艺优化过程漫长且生产给定量的单克隆抗体时间增加。
在治疗行业,确定单克隆抗体是否具有预期效果的早期研发工作通常使用体内来源的单克隆抗体进行,因为周转时间更短且生产成本更低。公司会同期开发体外生产工艺。产品开发和体外优化完成后,公司会生成最终产品有效性信息,并向FDA提交最终的工艺验证文件。治疗行业体外生产主要使用无血清体外技术,以避免反复接触外来抗原引起治疗相关的过敏反应。
而在诊断行业,激烈的竞争导致压倒一切的成本考虑,而外来抗原的存在则不那么重要。因此,通常使用体内来源的生产方式。体内程序经过优化,通过减少杂交瘤侵袭性和增加单克隆抗体分泌来提高生产率。这种优化可以将动物用量减少2~10倍,从而大大降低生产成本。
研究行业最关心单克隆抗体的生产周期和结合亲和力。使用体内方法还是体外方法取决于项目的目的以及该系统中细胞系产生的单克隆抗体的量。对于极小规模的生产,通常使用腹水生产,因为它比体外生产宽容得多,并且无需优化体外培养中的细胞系即可完成。
参考文献
[1]SUN Jing-jing, ZHOU Wei-wei, ZHOU Lei-ming, et al. Advance in Large-Scale Culture of Hybridoma Cells in Vitro[J]. China Biotechnology, 2018, 38(10): 82-89
[2]National Research Council (US) Committee on Methods of Producing Monoclonal Antibodies. Monoclonal Antibody Production. Washington (DC): National Academies Press (US); 1999. 5.
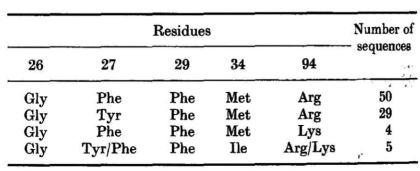
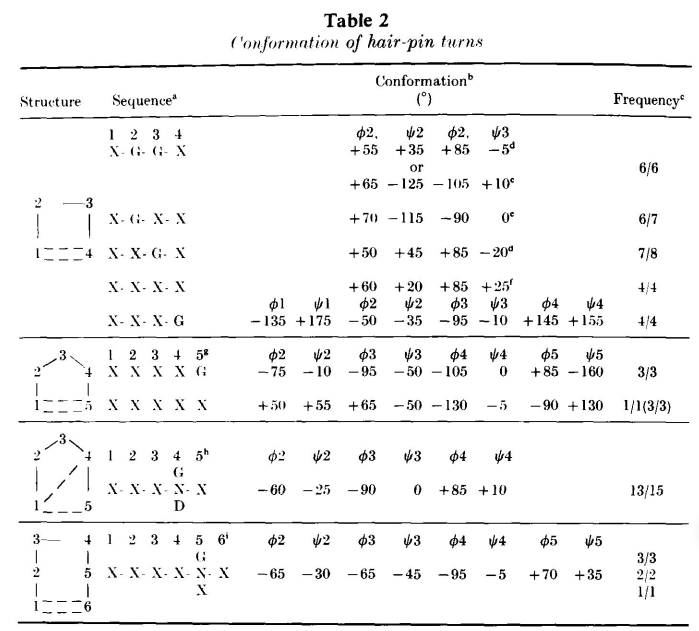
四个已知结构中的H1区长度相同,横跨于V区的顶部:
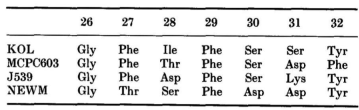
NEWM和MCPC603中H1的折叠非常相似,它们与KOL和J539中的折叠也很相似(图1)。
图1 VH结构域H1区构象
在观察到的H1结构中,位于26位的甘氨酸产生了一个角度较大的转角,其ϕ、φ值(+75, 0)超出了非甘氨酸残基所允许的范围。位于29位的苯丙氨酸深埋在框架区中,与残基34的侧链以及残基72和77的主链相邻。27位的残基(Phe或Thr)部分埋在残基94旁边的表面空腔中。在这四种结构中,位于26、34和94位的残基相同或相似,分别是Gly、Phe、Met/Tyr和Arg。
根据Kabat等列出的185个H1区已知的人类和小鼠VH区序列。在178个序列中,170个与已知结构中的序列长度相同,其余1个小鼠序列长一个残基,6个人类序列长两个残基。
在170个H1为7个残基的序列中,有115个94位残基是已知的。其中,四分之三在26、27、29、34和94位的残基与已知结构中的残基相同或非常接近:
环状结构的长度和H1与框架包装相关位点残基的保守意味着,至少在这些VH结构域中,H1的构象与已知结构中的构象接近。
H2区连接位于β-片层相邻链的框架残基52和残基56。在已知的VH结构中,H2环的长度各不相同:在NEWM中,它包含三个残基;在KOL和J539中,它包含四个残基;在MCPC603中,它包含六个残基。Kabat等列出了127个已知整个H2区序列的人类和小鼠VH序列。除一个序列外,所有序列中H2的长度都与已知结构之一相同:

42个H2区6残基的序列都属于小鼠的亚群Ⅲ。
NEWM H2区域的三个残基(Tyr53、His54和Gly55)构成了一个七残基转角的顶点。转角中的其他四个残基来源于框架区的一部分:
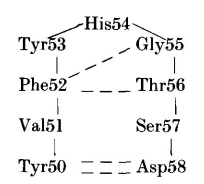
NEWM中发现的构象几乎和上文所述七残基转角的构象一致,在第五个位置(即NEWM中的第55位)有一个Gly,Asn或Asp。根据Kabat等列出的13个三残基H2区域中,有9个在55位有Gly残基,4个有Asp,预测这些H2区具有NEWM类似的构象。
J539和KOL中的H2区域(52a至55)形成四残基转角:

这些转角的构象由甘氨酸残基的位置决定。J539中的H2具有最常见的四残基转角构象:前三个残基处于近似αR构象,第四个残基(Gly55)处于αL构象。KOL中的H2则不同:Gly54处于αL构象,其他三个残基处于αR构象。
71个H2区为4个残基的序列,有10个Gly、Asn或Asp残基出现在54号位置,12个出现在55号位置,32个同时出现在54号和55号位置。在那些仅在54位有Gly、Asn或Asp残基的序列中,预测会出现类似于KOL中的H2构象。在55位有Gly的情况下,预测类似于J539中的H2构象。
MCPC603 H2区域的六个残基是十残基发夹转角的一部分。目前,实验和理论方面的证据不足,无法制定出这种大型转折的规则构象。因此不知道小鼠亚群Ⅲ中的其他六位残基H2区域是否与MCPC603中的构象接近。
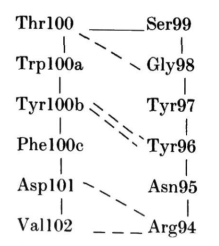
H3区域由残基96至101组成。VH结构由三个基因重组而成:VH(编码残基1至94或95)、D(编码1至13间残基)和JH。
人类的JH种系基因有6个,分别由以下氨基酸序列编码:

小鼠的JH种系基因有4个,由以下氨基酸序列编码:

由于D和JH基因的连接末端可能会发生变化,因此在JH基因开头编码的残基可能不会出现在最终结构中。体细胞突变会导致该区域序列的进一步多样性。因此,J539、NEWM、MCPC603和KOL的H3区域在长度(分别为6、7、9和15个残基)、序列和构象上存在很大差异也就不足为奇了。在此,我们将只讨论MCPC603的H3区,因为分析表明,其构象至少部分存在于其他几种免疫球蛋白中。
MCPC603中的H3区域形成了一个大的发夹环:
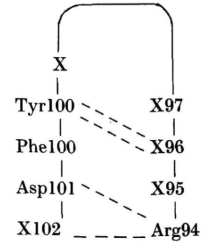
对于这种大环,ф、φ值范围将允许多种构象,而实际发现的构象将取决于与蛋白其余部分的堆积情况。在MCPC603中,H3的构象主要由VH结构域内和VL-VH界面的Arg94、Tyr100b、Phe100c和Asp101残基的相互作用决定。残基96至100a的侧链位于蛋白表面。
Arg94穿过H3发夹,与VL-VH界面的Asp101、Tyr100b和Phe100c形成表面盐桥(图2)。相当于100c位置上的残基通常是VL-VH界面保守核心的一部分,在83%的已知序列中,该位置的残基为Phe或Leu。在MCPC603中,Tyr100b与VL的Tyr49相邻,形成一个大空腔,两个Tyr残基的羟基都位于接触表面。在82%的VL结构域中,49为Tyr或Phe。相当于100b位置上的不同残基可产生不同的H3构象。例如,在KOL中是Gly,而与VL Tyr49相邻的空腔则由相当于100a位置的Phe填满。这使得KOL中H3的构象与MCPC603中的构象截然不同。
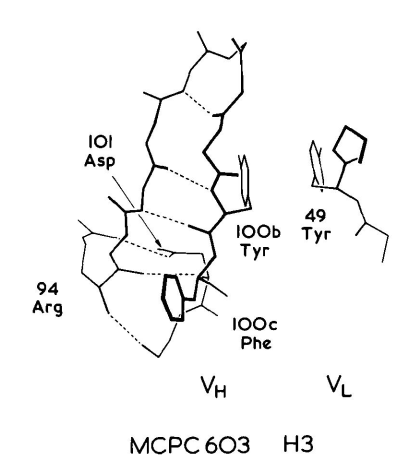
图2 MCPC603 H3区构象
位于100-100-101位的Tyr-Phe-Asp序列存在于人类基因JH2和JH4以及小鼠基因JH1和JH2中。人类JH5基因用Trp取代了Tyr。如果这些残基没有在基因重组或体细胞突变过程中丢失,通常认为这些J4基因会产生与MCPC603中的H3构象接近的H3构象。根据Kabat等序列表中查找出的长度至少为6个残基的H3区域,这些区域94位为Arg残基,101位为Asp残基,101位之前的两个位置有Tyr-Phe,即具有以下形式的区域:
根据Kabat等列出了的28个人类和77个小鼠序列的整个H3区域。其中,1个人类序列和48个小鼠序列完全符合长度和序列条件。另外5个人类序列很接近,它们的区别仅在于94位为Lys,Phe或Trp取代了Tyr,或Met取代了Phe。这54个序列中H3区域的长度分布:

在这些序列中,预测H3具有与MCPC603中相同的构象。
上文对超变区的描述表明,它们的主链构象完全由每个区域内的特定残基决定。实际上,这些构象可能会受到环境的影响。对特定区域的影响可分为两部分:构象的局部变化和结合位点中相对位置的变化。
衡量两种肽构象差异的标准是它们最佳叠加后原子位置的r.m.s.差异。在上面的章节中,我们报告了具有相同折叠的不同免疫球蛋白结构的超变区主链区域的r.m.s.差异。r.m.s.差异很小:在高分辨率下测定结构的r.m.s.差异小于0.5 Å,在中等分辨率下测定结构的r.m.s差异通常为1.0 Å或更小,这主要是由于肽段取向不同造成的。只有在H1区才会有显著差异,尽管构象上的差异仍然很小。
为了确定免疫球蛋白结构中超变环相对位置的差异,我们进行了以下计算:将Fab蛋白NEWM、MCPC603、KOL和J539 VL-VH框架残基拟合,将REI和RHE VL框架残基拟合。框架叠加后,我们计算了叠加同一折叠的超变区所需的额外位移,例如J539、REI和MCPC603的L1区的共同残基。
计算结果见图3。在Fab蛋白中,同一折叠的超变区在位置上相差0.2 Å至1.5 Å。出现这些差异的部分原因是,尽管VL-VH二聚体具有相同的残基接触模式和非常相似的堆积几何形状,但VH相对于VL的取向存在微小差异。
REI VL结构是通过Bence-Jonex蛋白确定的。它包含一个VL-VL二聚体,它们在REI中的排列与Fabs中的VL-VH排列非常相似。REI超变区相对于框架区的位置与Fabs中的位置相同。

图3 同源超变区相对于β-片层框架的位置差异
实线表示Fab结构中超变区之间的差异,断线表示Bence-Jonex蛋白和Fab超变区之间存在的差异。
因此,在这些结构中,超变区环境的不同只会导致主链构象的微小差异,其相对于框架的位置差异也不会超过1.5 Å。Bence-Jonex蛋白RHE和MCG中,超变异区的环境与免疫球蛋白中通常存在的环境截然不同。RHE中VL-VL二聚体的堆积方式与VL-VH二聚体的堆积方式截然不同,因此其超变异区的环境也与本文讨论的其他免疫球蛋白的环境截然不同。这些环境差异对RHE中L1、L2和L3的构象影响不大:它们与Fab KOL中的同源区的坐标r.m.s差小于0.3 Å。相对于框架区,它们确实会对L1、L2和L3位置产生一些影响:RHE中的位置与Fab中的位置相差可达2.2 Å。
在Bence-Jonex蛋白MCG的结构中观察到更复杂的情况,这种蛋白的晶体具有不对称单元组成的二聚体,两个VL单体处于不同的环境中。其中一个单体的L1区处于螺旋构象,与预期一致;而另一个单体的L1区则由于残基3至32与邻近分子的紧密接触而无法具有螺旋构象。密切接触产生无序结构表明,L1区域只有有限的灵活性。
上文中,我们将可变区的两个部分进行了精确的结构区分:保守的β-片层框架和主链可变区构象。这两部分对抗原结合位点有何贡献?
超变区聚集在VL-VH二聚体的一端,形成一个表面,其中一部分与抗原相互作用。图4显示了MOPC603中这一区域的空间填充图。在蛋白的这一部分,溶剂可接触到的VL残基为27至32、49至53和92至94,在VH中为28至33、52至56和96至100a。第3节中定义的β-片层以外区域在VL中为26至32、50至52和91至96,在VH中为26至32,52至56和96至101,溶剂可接触区域与其最多相差两个残基。在表6中,我们列出了J539、KOL和NEWM形成相同区域的溶剂可及残基。KOL、NEWM、J539和MCPC603中溶剂可及残基非常相似,但并不完全相同。环长度和序列的变化可能会导致环末端的一两个残基被掩盖或一两个框架残基暴露。
图4 MOPC603高变区空间填充图
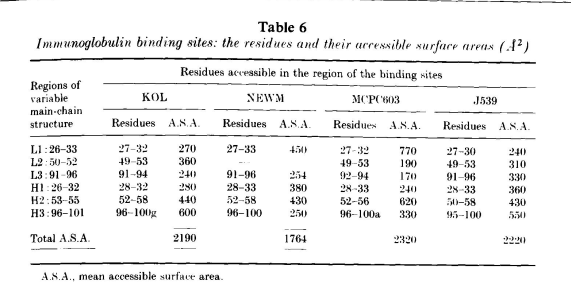
除H2外,表6中列出的区域与根据序列变异性确定的互补性决定区域(CDR)相似。表6中的H2包含残基50至58,相应的CDR包含残基50至65。Padlan发现,此CDR的前三个残基和后六个残基与NEWM和MCPC603具有相同的结构。残基59至65位于VH结构域的一侧,与其他超变区的距离相当远。残基59至65的侧链可以被溶剂接触到,其序列的变化可能仅仅反映了结构和功能约束的缺乏。
表6还列出了构成结合位点的各环状结构的可接触表面积。KOL中的H3不同寻常地大,有15个残基,对总表面积的贡献最大。而J539、NEWM或MCPC603中的情况并非如此,中等大小的H3区域与其他环的贡献相似。H3在抗体特异性中的重要作用不是来自于它的大小,而是来自于它在结合位点中的中心位置。
在J539、KOL和MCPC603中,超变异环总溶剂可接触表面积约为2250 Å2:而在NEWM中,由于未包含L2,其可接触表面积为1760 Å2。对寡聚蛋白的分析表明,在分离状态和结合状态的结构非常相似的情况下,稳定的结合是由表面积小于这些结合位点总表面积的表面形成的。通常情况下,单体有500 Å到1000 Å埋藏在界面中,占超变环可接触总表面积的四分之一到一半。这些界面中氢键和盐桥的数量各不相同。
对免疫球蛋白D1.3和鸡蛋蛋清溶菌酶形成的复合物的研究支持了抗体与蛋白之间的作用会涉及到与寡聚蛋白中的相似表面这一猜想。这种结合不涉及主链构象的重大变化。与溶菌酶接触的抗体残基在VL中为30、32、49至50、91至93,在VH中为30、32、52至54和96至99。互作界面由690 Å2的抗体表面和750 Å2的酶表面组成。
在这篇论文中我们试图识别决定高变区构象的残基。我们提出,如果我们定义的残基存在于其他免疫球蛋白序列中,那么它们的高变区将与已知结构的高变区具有相同的构象。对免疫球蛋白序列的分析表明,大多数高变区具有一组或多组主链构象。我们称这些构象为“典型结构”。
Vκ结构域AU和ROY的原子结构支持了我们分析的一些结论,这两种蛋白的序列与REI的序列分别有18和16个残基不同。其中8个位置的变化发生在高变区。

在30,31,93和96位的残基变化涉及体积和化学性质的巨大差异。然而,从上面给出的分析,我们认为它们应该产生与REI中相似的主链构象,事实上也是如此。
为了测试分析的准确性,我们用上述结论来预测新的免疫球蛋白的可变结构域。预测在通过X射线分析确定结构之前进行的。
基于本文所述工作的预测方法与前人所使用的方法有根本区别,之前的做法是将其免疫球蛋白高变区序列与已知结构中相应高变区序列进行了比较,然后从长度和总体序列同源性最接近的区域建立了每个环的模型。在某些情况下,调整是为了适应序列的差异。
基于本文所述工作的预测方法,如为了确定L1的构象,我们将检查第2、25、29、30、33和71位的残基。如果在这些位置上发现的残基与上文列出的一组残基相匹配,那么无论其他位置上的残基是什么,我们都会认为L1具有相应的典型结构。如果这些位置上的残基与其中一组残基不匹配,那么无论超变区其余部分的同源性多么接近,我们也无法鉴定为已知的典型结构之一。
我们对免疫球蛋白D1.3的结构进行了预测,并在确定其结构之前将预测结果发送给了进行X射线分析的小组。主链的构象是通过本文描述的分析方法预测的;对于侧链的构象,我们使用了之前描述的程序。在预测之后,我们根据2.8 Å电子密度图确定了D1.3的原子结构,并对框架结构和六个超变区中进行了预测。结果证实了晶体结构分析与预测已知结构相同。
D1.3的三个超变区(L1、L2和H2)与已知的典型结构长度相同,晶体学分析证实了这三个区域的折叠接近典型结构的预测。其他三个超变区的序列与已知的典型结构相同或长度相似,但在决定构象的位点上,它们的残基相似但不完全相同。在预测这些区域的结构时,我们必须判断这些差异是否会产生不同的主链构象。在其中的H3中,我们的预测是正确的,而在另外两个L3和H1中,我们的预测部分是错误的。
综上,我们确定了在已知结构中造成超变区构象的残基,如果这些特定残基出现在其他免疫球蛋白中,它们的超变区也将具有相同的结构。这也表明,当残基不完全相同时,即使变化很小,也很难有把握地预测结构。
我们对免疫球蛋白序列的分析表明,它们中许多超变区形成了已知结构的六个VL结构域或四个VH结构域中的典型结构之一。大多数超变区具有上述主链构象之一,这一结论对H3的适用可能有限,因为H3在长度和序列上的变化远大于其他区域。然而,我们的分析确实表明,在已知的小鼠序列中,有一半的H3区域的构象至少部分接近于MCPC603中的构象;D1.3中H3的成功预测证实了这一点。
对其他抗体晶体结构的分析将扩大典型结构的范围。尝试预测更多的结构,并在晶体学结构确定后对其进行测试,将提高我们理解负责其构象的残基可能发生的变化所产生的影响的能力。对抗体结构的预测不仅有助于测试我们对负责典型结构构象的残基的识别是否准确。它对于设计具有特定特异性的抗体也至关重要。
参考文献
Chothia,C. and Lesk,A.M. (1987) Canonical structures for the hypervariable regions of immunoglobulins. J. Mol. Biol., 196, 901–917.
生物药可能诱发适应性免疫反应,从而形成抗药抗体(ADA)。ADA的负面影响包括药物快速清除、药效丧失和过敏性休克。90%以上已批准的生物制剂会在研究的部分人群中诱发ADA。与原生人类蛋白相似性较低的产品诱导ADA的可能性会增加,其他因素也有可能影响ADA反应的发生,如配方、患者遗传学、患者病史和治疗方案。综上,ADA评价的目的有三个方面:了解药物疗效、药代动力学或不良事件是否与ADA的存在与否有关。例如,了解ADA是否会降低产品的循环浓度或血浆半衰期是一个重要的考虑因素,因为这些类型的ADA很可能会损害治疗药物的效果,并可能形成影响药物安全性的复合物。最后,了解ADA在治疗结果中的作用可为临床医生和研究人员提供设计或选择替代产品的机会,从而提高成功治疗的可能性。
ADA反应大致可分为两类:中和ADA(与药物产品结合并抑制药理功能)和非中和ADA(与药物结合但不直接抑制药物的理功能)。这两种类型的ADA都能通过诱导快速清除、阻止清除和增加生物利用度,或诱发其他免疫介导的结果(如细胞因子释放综合征、过敏反应或靶点耗竭)来改变药效。虽然不同药物和人群的ADA反应发生率差异很大,但检测灵敏度的提高已导致ADA检出率的增加,而且对其临床相关性的观察也各不相同。
ADA检测通常采用多层级的分析方法。首先使用高灵敏度筛选检测来确定样本中是否存在ADA,然后进行确证检测,以确定筛选检测中的阳性结果是否是由于特定的(可竞争的)相互作用所致。最后,进行滴度和/或功能检测(如中和、同种型、表位),以进一步确定ADA反应的特征并测量ADA反应的程度。在最初的检测试验中,可以采用各种测量蛋白-蛋白相互作用的方法,包括酶联免疫吸附试验(ELISA)、电化学发光(MesoScale Diagnostics)、微阵列(SQI Diagnostics)、固相(ImmunoCap)和珠基(Gyros, Luminex)试验。其他使用较少的方法包括表面等离子体共振、放射免疫沉淀和生物层干涉测量。无论采用哪种方法,灵敏度、对基质的耐受性以及循环治疗或治疗靶点的影响等因素都是需要考虑的重要因素。
评估和优化ADA检测方法其中一个挑战是阳性对照,通常是从免疫动物身上分离出来的单克隆抗独特型抗体,可以反映人类多克隆抗体反应的典型特征。虽然阳性对照通常不直接用于指定1级阳性阈值或切点,但它们在优化和描述检测性能方面的作用表明,了解不同阳性对照的表现,其对检测ADA的影响以及与研究结果之间的关系非常重要。阳性对照抗体克隆性、亲和力和靶标表位的差异都可能影响灵敏度,而阳性对照在循环药物存在时的功能又会进一步扰乱灵敏度。事实上,临床上越来越多地使用可延长血浆半衰期的抗体、可持续表达的新型基因递送方法以及其他新型长效注射技术,以延长治疗性抗体在循环中的存在时间。这种延长对ADA检测产生了影响,在开发检测方法时必须考虑到这一点。在药物存在的情况下,最广泛使用的ADA检测方法的信号可能会减弱或消失,从而导致假阴性结果。目前已开发出检测与治疗药物分离后的ADA的方法,但这些方法往往会对检测灵敏度、精确度和实用性造成影响。
ADA检测平台必须具有足够的灵敏度、稳健性和可重复性,才能在临床前及临床试验中发挥作用。采用多组药物和阳性对照在三种常用的检测平台ELISA,多重珠基免疫分析(MBA),电化学发光分析(ECL)进行性能检测评估,评估内容包括分析灵敏度(AS)或检测限(LOD)。各检测平台都采用桥连法进行检测,即抗体药物既用于固定ADA,又用于检测其存在。
检测药物选择临床阶段的六种HIV病毒特异性广谱中和治疗性抗体(10-1074,3BNC117,PGDM1400,PGT121,VRC01,VRC07)和一种双特异性治疗性抗体(10E8.4-iMab)。10E8.4-iMab和PGT121两种产品各有两种不同的抗独特型抗体,VRC01和VRC07两种产品共用一种抗独特型抗体。各平台抗独特型抗体与药物桥连的浓度依赖能力结果如图1所示。

图1 跨试剂和平台的ADA检测结果
垂直虚线表示FDA推荐的100 ng/mL灵敏度,MEI、OD和S/B分别用于MBA、ELISA和ECL检测。
在一种药物有多种抗独特型抗体的情况下,有时会观察到这些不同阳性对照抗体有不同的结合能力。虽然不同平台上识别同一种药物的抗体之间的差异基本一致,但也略有不同。例如,在ECL平台PGT121 ADA检测中,9E9和抗PGT121抗独特型抗体的AS相差三倍,而在MBA平台的检测中则相差九倍。在ELISA和ECL检测中,两种单克隆抗体的灵敏度相似,但峰值信号强度不同。相比之下,在MBA平台桥连检测中,10e8.4-iMab两株抗独特型抗体显示出相似的峰值信号,但抗iMab的AS几乎是抗10E8.4的十倍。
在各种平台中,ECL平台一般都具有最灵敏的ADA检测灵敏度和最大的动态范围。事实上,对于许多药物-抗独特型抗体配对,可能需要对ELISA和MBA方法进行大量优化,以达到建议的100ng/mL检测灵敏度。值得注意的是,在ECL平台中检测0%、25%和50%的集合正常人血清中的抗独特型抗体时,基质浓度对灵敏度的影响相对较小。总之,这些结果表明,不同的检测平台对相同试剂的灵敏度不同,不同的阳性对照对相同的实际实验检测的计算灵敏度也不同。
不同个体之间,甚至同一个体内随着时间的推移,血清中观察到的ADA反应的固有单价亲和力预计会有所不同。同样,抗独特型抗体对其目标药物的亲和力及其支持桥连(三分子)相互作用的倾向与支持二价分子内相互作用的倾向也会存在差异。研究人员检测了抗独特型抗体与相应药物的亲和力,以确定这一属性与ADA检测AS的关系。使用生物层干涉测量法(BLI)测量了二价和蛋白酶酶解的一价形式抗独特型抗体与相应药物的亲和力。药物与抗独特型抗体的二价亲和力介于<10 pM到1 nM之间,而一价亲和力介于1.54 nM到 39.7 nM之间。一价亲和力较高的药物-抗独特型抗体对的二价亲和力也较高(图2B)。虽然分子内和分子间的二价相互作用在BLI检测中无法区分,但在传统抗体中观察到的20-200倍的亲和力差异表明,药物和相应的抗独特型抗体之间的强结合很常见。相对的,双特异性抗体10E8.4-iMab不能实现分子内二价结合,其亲和力是酶解后双特异性抗体亲和力的五倍到八倍。利用这些数据,进一步研究了亲和力与ECL桥连AS测定结果之间的关系,结果发现无论是二价亲和力还是一价亲和力都与AS无关(图2C&D),这表明这一特性对于抗独特型抗体的选择并无帮助,高亲和力的试剂并不一定会导致高计算AS。对结合速率常数(ka)和解离速率常数(kd)与AS之间关系的分析发现,也无显著相关性。
10e8.4-iMab双抗的两种抗独特型抗体间,亲和力和AS的一致性并未被观察到:亲和力弱的抗独特型抗体,ADA检测AS优于亲和力强的(图2E)。相比之下,能识别两种VRCOx药物的5C9抗独特型抗体对VRC01的亲和力和AS均优于对VRC07(图2F)。
总之,虽然亲和力极弱的抗独特型抗体在用作阳性对照时肯定会影响计算出的检测AS,但抗独特型抗体制备过程中获得的亲和力数据可能并不能很好地预测检测敏感性。在确定哪些抗独特型抗体可以在最低浓度下被检测到方面,其他特性(如与药物架连的倾向性)可能同样或更为重要。
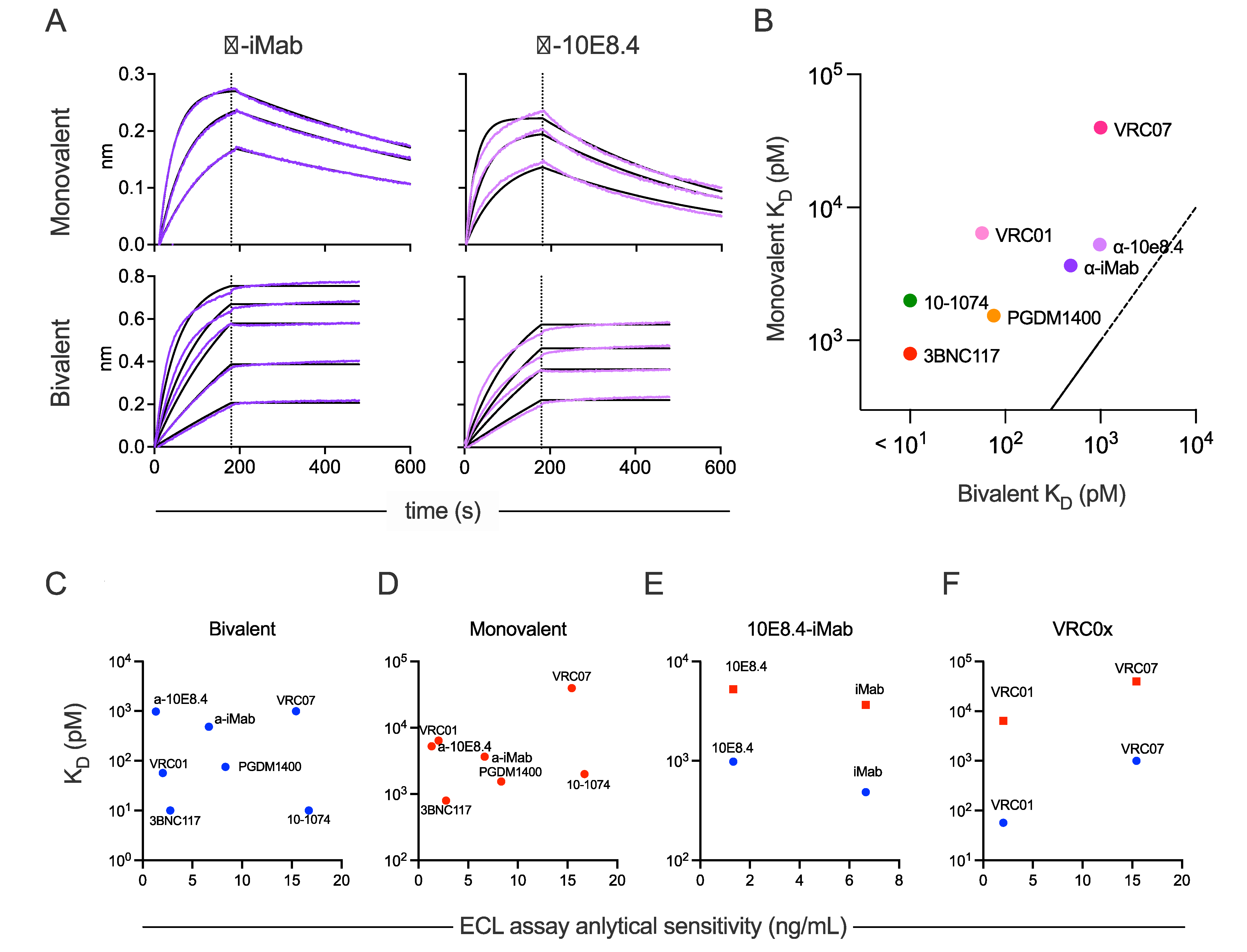
图2 药物-ADA的亲和力与分析敏感性之间的关系
除了在ADA检测中用作阳性对照外,抗独特型抗体对药物PK检测也至关重要,通常根据其在药物检测方面的灵敏性可靠性来选择。为了确定PK检测中选择抗独特型抗体时通常使用的数据是否会影响ADA检测的灵敏度,研究人员在标准筛选检测中对PGDM1400的七种抗独特型抗体进行了评估。在这组试剂中,有两个抗独特型抗体克隆因表现出高度相似的剂量反应曲线而被选中(图3A)。相反,在ECL ADA滴度检测中,这两株克隆的峰值信号相差两个数量级,AS相差一个数量级以上(图3B)。总体而言,在7个阳性对照中,没有观察到与传统ELISA和ECL ADA检测中信号的相关性(图3C)。这些发现共同表明,在抗独特型抗体筛选检测中具有高灵敏度的克隆在桥连法ADA检测中可能并不具有类似的高性能,这也进一步证实了阳性对照抗体选择对计算检测灵敏度的潜在影响。
除了用于确定检测灵敏度外,阳性对照还用于量化检测药物耐受性,并确保检测结果随时间的推移保持一致。研究人员利用PGDM1400的七种抗独特型抗体,研究了阳性对照选择对计算药物耐受性的影响。将单一浓度(400ng/mL)的PGDM1400抗独特型抗体与检测范围内的未标记药物预孵育,并在ECL ADA检测法中评估抗独特型抗体检测结果(图3D)。加入未标记药物会使阳性对照信号随剂量下降,加入250μg/mL未标记药物后,大多数阳性对照信号被完全抑制。计算了每种阳性对照的药物耐受性,并与没有未标记药物存在时的阳性对照信号(图3E)、LOD(图3F)和每种阳性对照的希尔斜率(图3G)进行了比较。剂量反应曲线的差异导致药物耐受性的不同,而药物耐受性与阳性对照的信号幅度、LOD或希尔斜率无关。这一观察结果表明,使用单一抗独特型抗体来定义ADA检测中对循环中存在药物的耐受性可能具有误导性,在解释药物耐受性值时应谨慎。使用多个阳性对照可对检测药物耐受性进行更有意义的评估,但与血清中存在的药物诱导ADA相比,任何阳性对照或一组对照都有可能表现出截然不同的药物耐受性。
图3 基于ELISA的抗独特型筛选与ADA检测结果的比较
由于在同一实际检测中依赖不同的抗独特型抗体导致计算出的AS存在差异,研究人员试图研究Fc结构域更保守的性质是否可以作为通用手段检测ADA灵敏度。可用于不同药物的阳性对照可作为检测灵敏度的通用衡量标准。在ECL ADA检测中评估了一组不同的多克隆和单克隆抗IgG二抗试剂,它们对抗体不同结构域具有特异性,且识别不同物种抗体Fc(图4A)。
相比抗独特型抗体在上述7种治疗性抗体中显示出较大的剂量反应分布,抗IgG多克隆抗体则更一致地显示出相对统一的结合模式。其余的差异可能来源于药物之间存在细微差异,或是化学标记和偶联制备过程中产生的差异。
而抗IgG单克隆抗体试剂显示出巨大的差异:识别位点在铰链区的试剂对部分药物基本没有观察到药物桥接,这表明单克隆抗体可能对药物之间的微小差异更敏感,只要这些差异属于它们识别的特定表位范围内。这些数据表明,可能没有真正通用的标准阳性对照,但对于治疗性抗体,仍有方法支持使用药物桥接ADA检测之间的基本比较。
最后,研究人员探究了阳性对照在确保检测性能一致性方面的作用。导致性能变化的最常见因素是药物偶联物的不稳定性,阳性对照的不稳定性对检测性能的影响并不常见。通过对两批VRC01药物偶联物进行了比较,这两批药物偶联物在制备时间上有所不同,但都使用了抗独特型抗体和多克隆山羊抗IgG试剂进行检测(图4B)。这两种阳性对照对药物偶联物随着制备时间的变化明显表现出不同的AS。同样,用浓度在线性范围内的七种抗独特型抗体和一种抗IgG阳性对照对新鲜制备和老化的PGDM1400药物偶联物进行了测试(图4C),老化药物偶联物在所有阳性对照中的检测信号都有所下降。然而,对于不同药物,药物偶联物制备时间的影响可能不尽相同,存在某些情况下AS没有变化,而另一些情况下信号完全消失(图4D)。这些观察结果表明,使用多组阳性对照抗体有助于识别药物偶联物中更微妙的变化。

图4 作为阳性对照替代品的非抗独特型抗体性能评估
比较三种不同平台的ADA桥接检测方法,包括ELISA、MBA和ECL,其中ECL的灵敏度最高。使用不同检测方法抗独特型抗体检测特征的差异导致了临床样本的ADA反应阳性率也会经常出现差异。随着更一致地使用桥接检测方法,可能会产生更有意义的比较分析。
评估ADA反应检测灵敏度缺乏统一标准,只能依赖性能各异的阳性对照来满足监管指南的要求。可用的阳性对照性能越差,计算出的检测灵敏度可能就越低,而如果因此必须检测浓缩血清,检测灵敏度实际上可能就越高。
对于抗独特型抗体亲和力属性和直接法ELISA检测AS预测桥接ADA检测AS和药物耐受性的能力,由于抗独特型抗体选择通常是根据药代动力学试验中使用的直接结合试验的灵敏度来进行的,因此很可能会导致ADA桥接试验灵敏度和药物耐受性不达标。同样阳性对照对药物偶联物的变化具有不同的敏感性,这可能会影响临床样本中ADA反应检测的可靠性。综上说明了在解释ADA检测结果时需要注意的事项,以及在检测开发和监测过程中纳入多个阳性对照的价值。
关于ADA检测阳性对照的进一步的建议:
1.用一组包含多个表位的不同阳性对照来定义灵敏度和药物耐受性,有助于建立更具代表性的检测特性,并确保检测结果的长期一致性。
2.在早期抗独特型抗体开发的过程中,同时考虑PK检测和ADA检测的应用,而不是在选定单个克隆后再进行ADA检测开发,这样可确保最适合PK检测但不适合ADA检测的抗独特型抗体不被优先考虑。
遗憾的是,目前似乎还没有实证检验的替代方法可以显示阳性对照敏感性、药物耐受性和识别试剂损伤的能力。不过,随着ADA检测越来越标准化,会进一步思考这些参数和优化选择对临床的影响,以支持对ADA检测数据进行最具临床相关性和影响力的解释。
参考文献
Weiner JA, Natarajan H, McIntosh CJ, et al. Selection of positive controls and their impact on anti-drug antibody assay performance. J Immunol Methods. 2024 May;528:113657.
免疫球蛋白的特异性由可变区高变区序列和长度决定,这些区域产生与抗原互补界面。先前研究比较了已知的高变区序列,发现在轻链的13个位点和重链的7个位点的残基是保守的。研究者认为,影响抗体结构的是这些保守残基位点,而不是整个高变区。这些残基在抗体中有固定的位置,可以用于结合位点的模型构建,以限制残基变化的位点的构象和位置。同时,对Fab和Bence-Jones蛋白结构分析的结果表明,长度相同但序列不同的高变区具有相同的主链构象。
基于以上认识,Chothia等通过对已知免疫球蛋白原子结构的分析,确定了已知结构共同的β-片层框架,确定了相对较少的残基,这些残基通过堆积、氢键或构成不寻常的φ、ψ或ω构象的能力,是高变区中观察到的主链构象的主要原因。这些残基一般存在于高变区和β-片层保守框架区。
如果上述残基存在于其他免疫球蛋白序列中,那么它们的高变区应与具有相同特征的已知结构高变区具有相同的构象,这些构象被称为“典型结构(canonical structures)”。如果在不影响特定规范结构构象的位点发生的序列变异,将会改变其呈现给抗原的表面。
研究基于Kabat等已经发表的一系列免疫球蛋白序列:对于轻链的可变区VL,包含大约200个完整序列和400个部分序列;对于重链的可变区VH,包含大约130个完整和200个部分序列。使用Kabat编号方案对这些序列进行编号,在少数情况下,某些高变区由结构叠加给出了不同于序列比对所建议的结果。
在表1中列出了已知结构的免疫球蛋白,其原子坐标可从蛋白质数据库(Protein Data Bank)获得,并提供了晶体图分析的参考。
VL和VH结构域具有同源的结构。每个结构域都包含两个大的β-片层,以主链间1 Å的距离相互包裹(1 Å=0.1 nm),以-30°的角度倾斜(图1)。每个结构域的β-片层由一个保守的二硫键连接。抗体结合位点由6个高变区形成,这些区域连接β-片层的β链。其中两个高变区是由不同β-片层的β链相连,另外四个是发夹弯——连接同一折叠中相邻两条β链的多肽(图2)。中小型发夹弯的构象主要取决于转角的长度和序列。表2总结了关于发夹弯构象的研究结果。
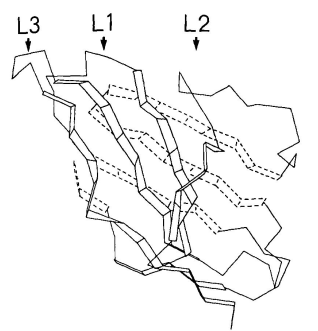
图1 免疫球蛋白可变区结构。
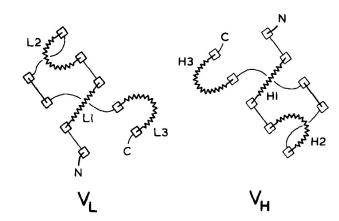
图2 免疫球蛋白高变区结合位点排列
不同免疫球蛋白分子的β-片层框架区非常相似。可变区框架区的结构相似性是由于形成域内部的残基趋向保守,以及内部残基总体量保守。通过对已知结构免疫球蛋白的主链原子进行最佳叠加比对(表1),计算同源残基原子位置的差异可以确定不同结构的相似性。图3给出了VL和VH β-片层框架的方案。β-片层框架包括VL残基4~6、9~13、19~25、33~49、53~55、61~76、84~90、97~107和VH残基3~12、17~25、33~52、56~60、68~82、88~95和102~112。
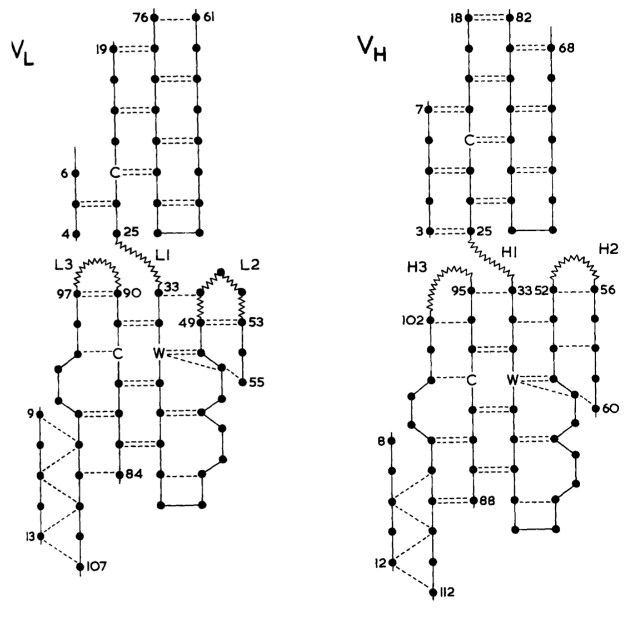
图3 β-片层框架折叠方案
框架的三级结构的主要决定因素是域内和域间的残基。表4列出了通常掩埋在VL和VH域中以及域间界面中的残基及它们的可及表面积。这些残基通常表现得较为保守。
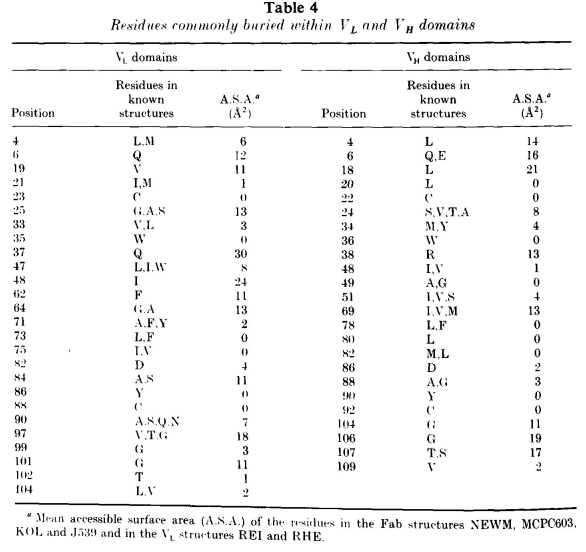
VL序列中有两个位点的保守残基性质取决于链的类型。Vλ 71位和90位残基通常分别为Ala和Ser/Ala;而Vκ相应残基通常为Tyr/Phe和Gln/Asn。这些残基与高变区相连,并对环构象产生一定的影响。
框架结构的保守性扩展到紧邻高变区的残基。若对一对免疫球蛋白分子的框架区进行比对,这些残基位置的差异在大多数情况下小于1 Å(表5)。相反,在与框架区相邻的高变区残基,位置可以相差3 Å或更多。
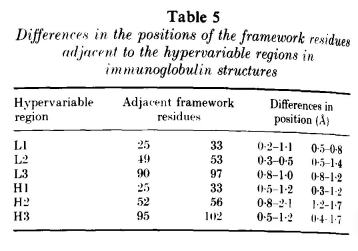
6个主链构象不同的环状结构是抗体结合位点的一部分,分别由VL结构域L1、L2、L3于26~32、50~52和91~96位点和VH结构域H1、H2和H3的26~32、53~55和96~101位点组成,与Kabat等根据序列变异性定义的互补决定区有些不同:VL的24~34、50~56和89~97,VH的残基31~35、50~65和95~102。
在已知的VL结构中,L1区残基26至32的构象展示了轻链的类别特征。在Vλ结构域中,构象呈螺旋状,而在Vκ结构域中,构象呈延伸状。这些构象差异是L1区和框架的序列差异造成的。
RHE和KOL的L1区包含9个残基,分别为26至30、30a、30b、31至32,NEWM多一个残基。RHE和KOL的L1区具有相同的构象:它们的主链原子位置仅有0.28 Å的均方根(r.m.s.)差异。将NEWM的L1区与KOL和RHE的L1区叠加显示,额外的残基插入在残基30b和31之间,对该区域其余部分的构象影响很小;将NEWM中26至30b和31至32的主链原子与KOL和RHE中26至32的主链原子叠加,分别有0.96 Å和1.25 Å的均方根差异。通过结构叠加得出的KOL、RHE和NEWM Vλ L1区的序列比对如下:

在这三种结构中,残基26的羰基和残基29的酰胺之间形成氢键Ⅰ型转角。残基27至30b形成一个不规则的螺旋(图4),该螺旋位于β-片层核心的顶部。残基30的侧链深入核心,占据了残基25、33和71之间的空腔。在观察到的结构中,决定L1构象的主要因素是残基25、30、33和71的堆积。Vλ RHE、KOL和NEWM在这些位点具有相同的残基:Gly25、Ile30、Val33和Ala71。(L1残基Asp29或Asn29因与L3接触而被掩埋)。
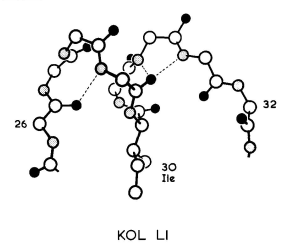
图4 KOL Vλ结构域L1区构象
根据Kabat等列出的33个L1区序列已知的人类Vλ结构域,其中Ⅰ、Ⅱ、Ⅴ和Ⅵ亚组中的21个序列的L1区长度与RHE、KOL或NEWM中的序列相同。其中18个序列保留了上述构象的关键残基,这意味着这18个L1区的构象与RHE、KOL或NEWM中的构象相同:

而亚群Ⅲ和Ⅳ中有13个L1区序列已知,长度比RHE和KOL以及其他Vλ亚群的序列更短,保守残基模式也大不相同。
根据Kabat等列出的29个L1区序列已知的小鼠Vλ结构域。这些L1区的长度与NEWM中的相同,它们的保守残基模式也与KOL/NEWM中的相似,但不完全相同:Ser25,Val30,Ala33,Ala71。
图5展示了三个已知的Vκ结构:J539、REI和MCPC603中L1区的构象。J539的L1有6个残基,REI有7个,MCPC603有13个。J539的L1区具有伸展构象(extended conformation)。REI中残基26至28具有伸展构象,残基29至32形成扭曲的Ⅱ型转角。
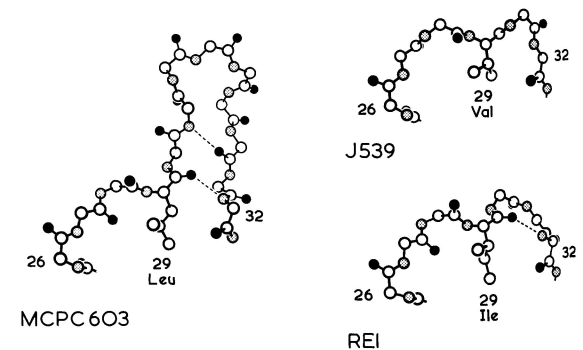
图5 Vκ结构域L1区构象
MCPC603中的另外6个残基都出现在这一转角区域。在这三种结构中,残基26至29和32的主链具有相同的构象。对J539、REI和MCPC603中这些残基进行叠加,得出的位置r.m.s.差异为0.47 Å至1.03 Å。结构叠加所得出的序列比对结果为:
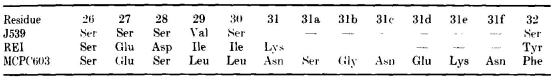
在J539、REI和MCPC603中,残基26至29延伸至整个β-片层框架的顶部,其中一个残基29被掩埋在其中,残基29的主要接触点是残基2、25、33和71。残基29对框架内部的渗透不如残基30在Vλ结构域中的渗透大,Vκ结构域中存在的深腔被残基71的大侧链填满。在J539、REI和MCPC603中,参与L1堆积的残基(2、25、29、33和71)非常相似,分别是Ile、Ala/Ser、Val/Ile/Leu、Leu和Tyr/Phe。
MCPC603中的6个残基30至30f形成了一个发夹环,从结构域中延伸出来,没有良好有序的构象。
Kabat等曾指出,当时已知的Vκ序列L1区某些位置上的残基是保守的,并认为这些残基具有结构性作用。上述对Vκ结构的分析以及目前已知的大量序列中残基保守模式证实了残基25、29和33的结构作用。
根据Kabat等列出的65个人类Vκ序列和164个小鼠Vκ序列,有59个人类序列和148个小鼠序列在涉及L1区包装的位点上的残基与已知结构中的残基非常相似:

这些序列中L1区的残基数量各不相同:
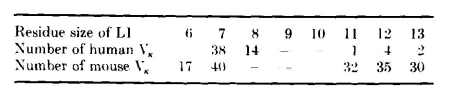
掩埋在L1区和框架之间位置的残基的保守性意味着,在绝大多数Vκ结构域中,残基26至29的构象与已知结构中的构象接近,其余残基如果数量少,则形成一个转角,如果数量多,则形成一个发夹环。
在已知结构中,L2区具有相同的构象。L2结构的相似性源于三残基转角的构象要求和L2包裹的框架区残基的保守。已知结构的L2区由三个残基组成,50至52:

这三个残基连接框架β-片层中两条相邻的链。残基49和53以氢键连接,L2区形成由三个残基组成的发夹转角。
这五种结构L2区的构象非常相似:其主链原子位置的r.m.s.差值在0.1 Å到0.97 Å之间。构象间的唯一差异在于残基50和51之间的肽的方向。在MCPC603中,这种差异与50位的甘氨酸残基有关(图6)。
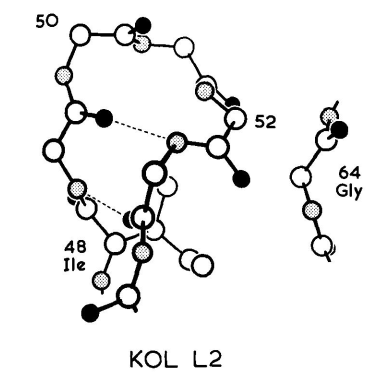
图6 KOL Vλ结构域L2区构象
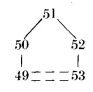
L3区域(残基91至96)是两条相邻的β-片层链之间的连接区域。对该区域已知结构和序列的分析表明,绝大多数κ链拥有共同构象,且与λ链的构象截然不同。
NEWM Vλ L3区有6个残基,KOL和RHE有8个残基:

在所有三种Vλ结构中,残基91至92和95至96构成了β-片层框架结构的延伸,残基92和95之间存在主链氢键:
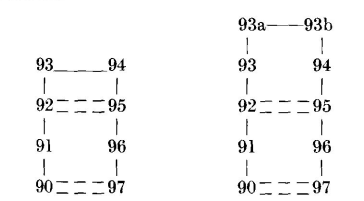
NEWM中残基93和94形成双残基Ⅱ型转角。RHE和KOL中的残基93、93a、93b和94形成了相同构象的四残基转角:它们的主链原子位置的r.m.s.差异为0.19 Å。与KOL和RHE一样,四残基转角几乎在所有转角的第四个位置(即此处的第94位)为Gly或Asn。
根据Kabat等列出的27个人类Vλ结构域和25个小鼠Vλ结构域(L3区序列已知),这些序列中L3区长度的分布情况如下:
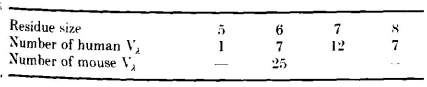
六残基的L3区中,预测会像NEWM一样,将91至92和95至96连接到框架β-片层上,而93至94形成一个双残基发夹转角。同样,在具有八个残基的L3区域中,预测91至92和95至96延续β-片层框架,而93、93a、93b和94将形成四残基转角。
REI、MCPC603和J539中的L3区长度相同:
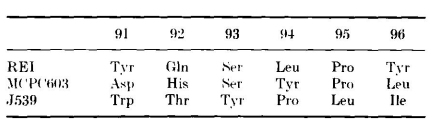
REI和MPCPC603 L3区具有相同的构象:残基91至96的主链原子位置的r.m.s.差异为0.43 Å。而J539中的L3与REI和MCPC603中的L3区具有不同的构象。
通常,对于六残基环,可能会预测残基92和95的主链原子形成氢键,残基93和94形成转角(同Vλ)。而在REI和MCPC603中,这种构象被位于95位的脯氨酸残基所阻止。在这两种Vκ结构域中,残基92具有αL构象,而Pro95具有顺式肽。这使得残基93至96处于伸展构象中(图7)。这种特殊的L3构象的重要决定因素是框架残基90的侧链与其主链原子形成的氢键。虽然90位的侧链不尽相同(REI的侧链是Gln,MCPC603的侧链是Asn),但酰胺的位置相同,作用也相同:NH基团与93和95的羰基形成氢键,O原子与92的酰胺形成氢键(图7)。
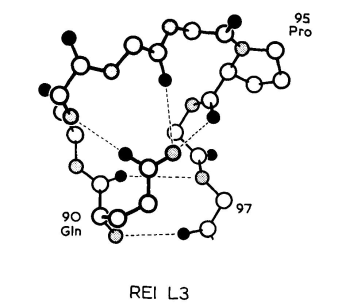
图7 REI Vκ结构域L3区构象
虽然J539中的L3长度为6个残基,但它在95位的残基是Leu而不是Pro,形成了一个两残基的发夹转角:
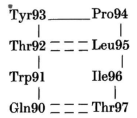
由于在94位有Pro残基,这种转角的构象与Vλ链的不同:Tyr93的ф、φ值为-51°,+131°;Pro94为顺式肽,ф、φ值为-46°,-54°。
Kabat等发现,Vκ序列L3区90和95位的残基是保守的,并认为它们具有结构作用。根据Kabat等列出的121个已知整个L3区序列的人类和小鼠Vκ结构域,L3区域的长度分布如下:
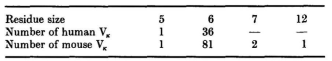
在117个含有6个残基的L3区序列中,93个的95位为Pro,90位为Gln或Asn。它们的长度和序列相同意味着这93个的L3区构象与REI和MCPC603中的相同。还有16个在94位有Pro,但在95位没有,它们很可能具有在J539中发现的L3区构象。
参考文献
Chothia,C. and Lesk,A.M. (1987) Canonical structures for the hypervariable regions of immunoglobulins. J. Mol. Biol., 196, 901–917.
流式细胞术(flow cytometry,FCM)是利用流式细胞仪对液流中排成单列的细胞或其它生物微粒(例如微球,细菌,小型模式生物等)逐个进行快速定量分析和分选的技术,具有高通量、高灵敏度和定量分析三大优势。流式细胞仪可快速测定库尔特电阻、荧光、光散射和光吸收来定量测定细胞DNA含量、细胞体积、蛋白质含量、酶活性、细胞膜受体和表面抗原等许多重要参数。根据这些参数将不同性质的细胞分开,以获得供生物学和医学研究用的纯细胞群体。
流式细胞仪主要由三大系统组成:液路系统、光学系统和电子系统。
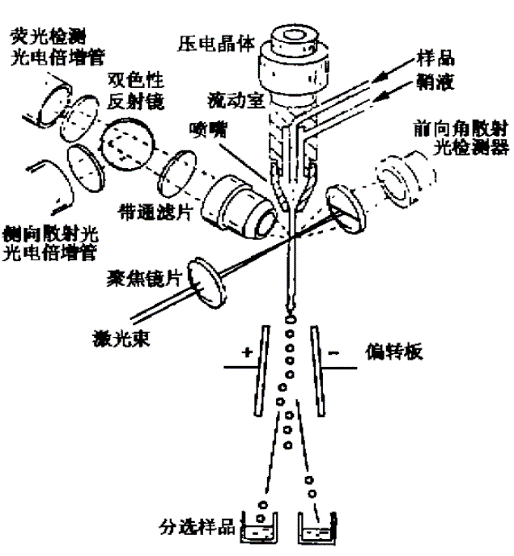
图1 流式细胞仪示意图
液路系统由进样管、鞘液、流动室、压力控制系统和废液回收系统组成。液路系统可以将样品排列成单列细胞/颗粒流,并使细胞/颗粒稳定高速的通过流式细胞仪的检测点。光学系统由激发光源、光信号收集、分离系统(二向色镜、滤光片)和光信号检测器组成。当细胞/颗粒通过检测点时,激光束照射到单个细胞/颗粒上,产生前向角散射(FSC)和侧向角散射(SSC)和荧光。所有这些光被透镜收集,二向色镜和滤光片分离并导向相应的检测器,在检测器内转化为光电流。电子系统由信号放大器和模数转化器组成。电子系统对收集的信号进行放大,通过模数转化器转化为数字信号,以备保存和后续分析或下一步操作(分选)。
待测样品经荧光染料染色或标记后制成样品悬液,通过进样管进入流动室,由喷嘴喷出而成为细胞液流下并与入射激光束相交。细胞被激发而产生散射光和激发荧光。散射光信号包括前向散射信号和侧向散射信号。散射光信号和荧光信号被光电二极管和光电倍增管接收后可转换成电脉冲信号。无论是散射光或荧光信号,检测的目的都是要分析不同的粒子,分析的方法有两种,一是测量粒子通过激光束的最大电压,二是测量脉冲的面积,最后再经过A/D转换器将脉冲信号变换成二进制数字信号由计算机处理。散射光信号基本上反映了细胞体积的大小,荧光信号的强度代表了所测细胞膜表面抗原的强度或其核内物质的浓度。粒子被逐一分析后即在屏幕上显示各种图形:直方图、二维散点图、等高图、灰度图和三维立体图等。
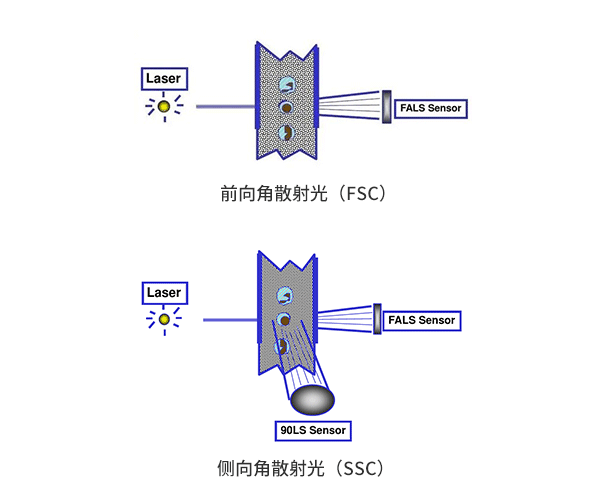
图2 前向角散射(FSC)和侧向角散射(SSC)
由超声振荡器产生高频振荡,使流动室发生振动,把喷嘴喷出的细胞液流断裂成一连串的均匀小液滴,有的液滴含有细胞。这些细胞在形成液滴前,光学系统已经测定了它们的信号(代表细胞的性质),如果测得信号与所选定的要进行分选的细胞性质符合,仪器给充以短暂的正或负电荷。当该液滴离开液流后,其中被选定细胞的液滴就带有电荷,而不被选定的细胞液滴则不带电。带有正电或负电的液滴通过高压偏转板时发生向阴极或者向阳极偏转,从而达到分类收集细胞的目的。
一般在硏究细胞样品时,首先关注的就是样品中细胞的FSC-SSC散点图,x轴代表FSC值,y轴代表SSC值,将细胞都显示于该散点图中。FSC的值代表细胞的大小。细胞体积越大,其FSC值就越大。SSC的值代表细胞的颗粒度(granularity)。细胞越不规则,细胞表面的突起越多,细胞内能够引起激光散射的细胞器或者颗粒性物质越多,其SSC值就越大。
图3样品中的细胞包含淋巴细胞、单核细胞、粒细胞等。在该图中,FSC越往右,其信号越强,说明细胞越大,单核细胞和粒细胞的体积大于淋巴细胞。SSC越往上,说明细胞颗粒度越强,单核细胞和粒细胞颗粒度比淋巴细胞强,因此可以根据细胞的大小和颗粒度实现了对细胞进行分群和分类。
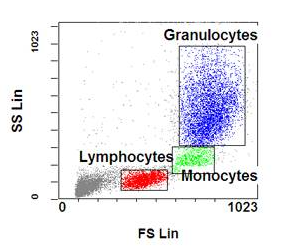
图3 流式细胞散射光散点图
流式细胞散点图在表示荧光信号的时候也有非常重要的作用。调节性T细胞的有两个标志CD4和CD5。如图4左图所示,X轴表示CD4信号,Y轴表示CD25信号。由CD4、CD5的丰度可以将这些细胞分为CD4-CD25+(左上),CD4-CD25-(左下),CD4+CD25(右上),CD4+CD25-(右下)四个部分。
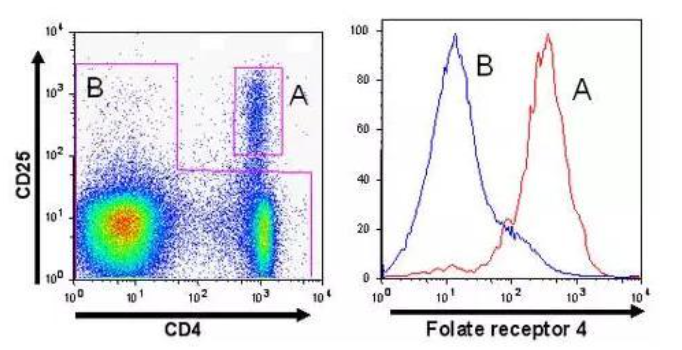
图4 流式细胞荧光散点图和直方图
图4右图则是相应的直方图,x轴表示相对荧光强度,y轴表示相对细胞数量。右图中红色的线对应的是左图的A门,蓝色的线对应的是左图的B门。散点图和直方图的灵活组合可以展示很多信息。
1.免疫分型
2.细胞凋亡检测
3.细胞周期检测
4.可溶性蛋白检测






南京德泰生物工程有限公司 Nanjing Detai Bioengineering Co.,Ltd. ©2025 All Rights Reserved
 苏公网安备32011202001300
苏公网安备32011202001300