
蛋白质相互作用的研究是阐释基因功能的重要手段,近年来发展起来的高分辨率质谱技术为蛋白质复合体的鉴定提供了强有力的工具,因此确定蛋白质间相互作用的限制因素不是蛋白质鉴定,而是蛋白质复合体的纯化。传统的纯化方法(如亲和层析或免疫共沉淀)难以得到接近天然状态的蛋白复合体,且实验结果可能存在假阳性。
串联亲和纯化(tandem affinity purification,TAP)是一种能快速研究体内蛋白相互作用的技术,经过两步特异性亲和纯化,可快速得到生理条件下与靶蛋白质存在真实相互作用的蛋白质。TAP方法最初用于酵母中,因其具有通用性、高效性、高纯度及假阳性低等特点得到了快速发展,至今已成功运用于许多其他生物(哺乳动物、植物等)相互作用的研究。
强大、灵敏度高的高通量质谱(MS) 技术的出现,可以在飞摩尔(fm)以下的范围检测肽分子,常用来鉴定相互作用的蛋白质复合体或复合体亚基,大大促进了细胞蛋白质组的生物化学纯化方法的发展。但通常难以获得足够量纯化的蛋白质复合体,成为质谱在这方面应用的一个限速步骤。1999年Rigaut 等共同提出了一套分离复合蛋白的新方法–串联亲和纯化(Tandem affinity purification,TAP), 兼具标准亲和纯化和免疫共沉淀两种生化方法的优点,为蛋白质复合体的分离鉴定提供了一条新途径,串联亲和纯化联合蛋白质质谱(MS) 鉴定技术可以进行生物化学纯化,双分子和大分子蛋白质复合体的分析
利用串联亲和纯化检测蛋白质间相互作用的原理是在靶蛋白质一端嵌入一个特殊的蛋白标签(TAP tag),不破坏靶蛋白质调控序列。将构建好的靶蛋白质粒转入靶细胞内进行表达,如果靶细胞内存在相互作用蛋白,则会与靶蛋白结合形成蛋白复合物。利用TAP标签进行两步连续的亲和纯化获得接近天然条件的靶蛋白复合物。随后用质谱等技术对靶蛋白复合物进行鉴定,从而达到研究蛋白质间相互作用的目的。

最初设计的TAP标签主要由金黄色葡萄球菌蛋白A的两个IgG结合域(ProtA)和一个钙调蛋白结合肽(calmodulin-binding peptide,CBP))构成,中间被一个TEV蛋白酶的酶切位点隔开(见右图)。
用IgG亲和柱进行亲和纯化,ProtA与亲和柱紧密结合,洗去未结合的杂蛋白。再用TEV蛋白酶酶切,获得CBP-融合蛋白。
将酶切后获得的CBP-融合蛋白与偶联有钙调蛋白的亲和柱混合进行第二轮亲和纯化。在钙离子存在下,CBP就会与钙调蛋白紧密结合,用含有EGTA的洗脱液进行温和洗脱,即可得到高纯度的靶蛋白(如果靶细胞内存在相互作用蛋白,则获会得靶蛋白复合物)。
两轮的亲和纯化顺序为先利用IgG亲和柱进行纯化,再利用钙调蛋白偶联亲和柱进行纯化。如果纯化的顺序互换,可能存在TEV酶污染的问题。

图2:串联亲和纯化(TAP)原理
构建TAP标签-蛋白质粒导入到细胞或生物体内。获得可正常表达的融合TAP标签蛋白。
转染后,获得细胞抽提物。需要注意的是:各无论何种抽提策略,都必须保证能够降低非特异性相互作用的干扰,同时尽可能不要破坏自然存在的相互作用。
随着TAP技术的发展,越来越多的标签供不同串联组合,常见的TAP标签有FLAG 标签、金黄色葡萄球菌蛋白 A 的 2个 IgG 结合域(ProtA)、Strep 标签、His 标签、钙调蛋白结合肽(calmodulin2binding peptide,CBP) 以及角质素结合结构域(chitin2binding domain,CBD)等。在选择串联标签时应综合考虑纯化回收率、纯度、对融合蛋白的结构和生物学的影响以及成本等因素。
在两亲和纯化标签之间存在酶切位点,该位点以烟草蚀纹病毒酶切点(TEV)最为常见,原因是TEV蛋白酶高效特异,基本不含其它细胞蛋白识别点,使用TEV蛋白酶切掉相关蛋白质的概率极低。
标签融合的位置: 融合标签可以加在靶蛋白的N端也可以加在靶蛋白的C端,通常推荐使用C-端TAP标签,这样可以使融合蛋白的表达在天然启动子的控制下,蛋白表达不受影响(有的蛋白当其C端加上其它肽段后会影响该蛋白的功能,此时应利用N端TAP标签)。如果不知如何选择标签,可以在实验初始设计两条融合蛋白(标签位置一条在N端,一条在C端),以应对无法顺利表达的问题。
酵母双杂交系统检测蛋白相互作用
免疫荧光共振检测蛋白相互作用
传统的相互作用分析方法(如GST-pull down、免疫共沉淀、酵母双杂交技术等)在大规模研究蛋白相互作用方面有着一定的局限性。随着生物芯片技术的发展,蛋白质芯片技术被应用于蛋白间相互作用研究。蛋白质芯片技术能够快速、高通量的进行蛋白质相互作用的研究。
蛋白质芯片技术的产生得益于生物芯片技术的发展。1991年美国Affymetrix公司成功研制了世界上第一块DNA芯片,从此拉开了研究生物芯片技术的帷幕。DNA芯片可以应用于对生物样品中的各种已知/未知核酸序列的表达进行检测和比较,但DNA芯片在对蛋白质功能结构的研究方面有很大的限制。为了进一步对蛋白质的功能与结构进行研究,蛋白芯片技术应运而生。
蛋白质芯片又称蛋白质阵列或蛋白质微阵列,是一种体外检测相互作用的方法。其基本原理是以蛋白质分子作为配基(探针蛋白),将其有序地固定在固相载体(滴定板、滤膜、玻璃片等)的表面形成微阵列;用带有荧光标记的蛋白质(或其它分子)与之作用,经漂洗将未结合的成分洗去,经荧光扫描等监测方式测定芯片上各点的荧光强度。通过荧光强度分析蛋白质与蛋白质之间相互作用的关系,由此达到测定各种蛋白质功能的目的。

蛋白芯片上的蛋白根据研究目的的不同,可以选用抗体、抗原、受体、酶等具有生物活性的蛋白质,利用原位制备或制备后交联等方法,将不同的探针蛋白质,按设计好的序列固化于固相载体。蛋白芯片制备的关键是制备时注意维持蛋白质的天然构象(防止蛋白变性,维持原有特定的生物学活性)。
蛋白质芯片的探针蛋白特异性高、亲和力强,受其它杂质的影响低,因此对生物样品的要求较低,只需对少量样本进行沉降分离和标记后,即可加于芯片上进行分析和检测。甚至可以直接利用生物材料(血样、尿液、细胞及组织等)进行分析。
目前主流蛋白质芯片的信号检测方式主要是荧光检测形体系。将蛋白质芯片与荧光素标记的生物靶分子(核酸或蛋白质等)进行杂交,洗脱未结合组分后通过共聚焦荧光扫描仪或CCD荧光成像仪在特定的波长下激发荧光,获得反应结合的信号。
对荧光标记的芯片用共聚焦荧光显微镜进行扫描,然后通过计算机分析出每个点的平均荧光密度。
在每个芯片的制作过程中应设计有阴阳性对照反应,或已在多次实验中找到一个判断阴阳性结果的界值,作为判断结果的根据。将每个点的荧光密度或灰度除去背景干扰后与相对界值进行比较,根据信号的有无、多少进行定性或定量分析。当然,在结果分析过程中需要对大量数据进行复杂处理,这些都需要通过特定的计算机软件来完成。
利用蛋白质芯片技术可以检测蛋白-蛋白、蛋白-小分子、蛋白-DNA、蛋白-抗体、蛋白-脂类之间的相互作用。

Far-western blot是一种检测蛋白间相互作用的分子生物学方法。它可以验证已知蛋白间的相互作用,或分析已知蛋白和未知蛋白间的相互作用。Far-Western blot技术与Western blot相似。在Western blot实验中,用特异性抗体(一抗)去检测膜上的蛋白,HRP标记的二抗与一抗结合,通过显影观察膜上的蛋白。而在Far western blot中,将靶蛋白固定在PVDF/NC膜上,用诱饵蛋白(已知蛋白)作为探针去检测膜上的靶蛋白,再利用特异性抗体孵育、检测,以此来分析靶蛋白和诱饵蛋白间的相互作用。
Far-western blot实验流程主要包括:①凝胶电泳;②转膜;③封闭;⑥孵育;⑦检测。下面对Far-western blot的实验操作流程做系统介绍。

通过凝胶电泳将样品中不同大小的蛋白分离开来。电泳可以用十二烷基硫酸钠 – 聚丙烯酰胺凝胶电泳SDS-PAGE或天然PAGE分离。
样品中的蛋白在凝胶上分离后,将蛋白质从凝胶转移到膜上。此步骤的目的为将蛋白附着到膜的表面,使蛋白变得易于探测。在转膜的过程中需小心操作,避免污染问题。如果转膜过程存在污染,则最终检测结果会出现很高的背景。
转膜结束后一般采用异源性蛋白质封闭整张膜,以阻塞非特异性结合位点。常用的封闭剂有脱脂奶粉、BSA等。需要说明的是,封闭剂有可能会发生交叉反应或者以其它的方式破坏待研究的蛋白相互作用,需要根据经验确定合适的封闭剂。
将诱饵蛋白与封闭后的膜共孵育,使诱饵蛋白与NC膜上相互作用的蛋白结合,孵育后洗去未结合的诱饵蛋白。探针蛋白通常可以利用大肠杆菌表达系统生产出来,虽然细胞裂解液也可以作为探针蛋白,但是为了降低实验背景,最好选择纯化的蛋白作为探针(诱饵蛋白的纯度越高,实验的成功率就越高)。
检测探针蛋白的策略通常有以下几种:
为了提高结果的准确性,需要设置合适的实验对照。例如,如果诱饵蛋白为GST-融合蛋白,则单独设置一组GST标签实验组作为阴性对照,排除GST标签本身与膜上的靶蛋白存在非特异性结合的可能性。
| 对比项目 | Far-western blot | Western blot |
|---|---|---|
| 凝胶电泳 | 天然(通常)或变性 | 天然或变性(通常) |
| 转膜 | PVDF膜/NC膜 | PVDF膜/NC膜 |
| 封闭 | 脱脂奶粉/BSA | 脱脂奶粉/BSA |
| 检测策略 |
|
一抗→酶标二抗 |
| 实验对照 | 需要 | 不需要 |
注意事项:
检测标记时,通常用酶(辣根过氧化物酶或碱性磷酸酶)标记抗体。相比之下,诱饵蛋白不选用酶标记,因为大的酶标记可能在空间上阻碍诱饵和靶蛋白之间结合。
酵母双杂交系统,又称蛋白阱捕获系统,是由Fields和 Song等人根据真核转录调控的特点创建的。利用酵母双杂交系统能够快速、直接分析已知蛋白之间的相互作用,并能寻找、分离与已知蛋白相互作用的配体,在研究抗原和抗体相互作用、发现新的蛋白质和发现蛋白质的新功能、筛选药物作用位点及药物对蛋白互作影响、建立基因组蛋白连锁图等方面应用广泛。
酵母双杂交系统的建立是基于对真核生物转录调控过程的认识。真核生物中基因转录需要转录激活因子的参与,真核生物的转录激活因子含有两个不同的结构域:DNA结合结构域(DNA binding domain,DNA-BD)和DNA转录激活结构域(Activation domain,AD),这两个结构域可以独立分开,功能互不影响。BD和AD分别单独作用并不能激活转录反应,只有当二者在空间上充分接近时,才呈现完整的转录激活因子活性,使下游基因得到转录。根据这一原理,可设计酵母双杂交系统。
将两待研究蛋白(蛋白X与蛋白Y)分别与BD、AD结构域构建融合质粒。将构建好的两个质粒转入同一酵母细胞中表达,如果两蛋白之间不存在相互作用,则下游基因(报告基因)不会转录表达;如果两个蛋白存在相互作用,则BD与AD两结构域空间上很接近,从而下游基因(报告基因)得到转录。判断通过报告基因表达与否,即可判断两蛋白之间是否存在相互作用。
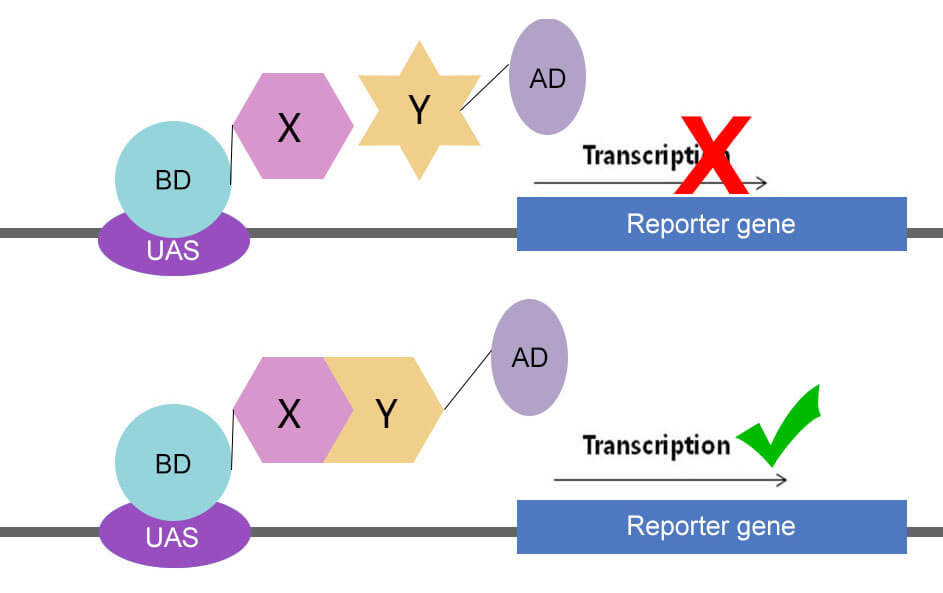
图1:酵母双杂交原理
报告基因作为一种营养标记,常用的有HIS3,URA3,LacZ和ADE2等,对应的宿主菌则是相应标记的缺陷型细胞,必须要在含有该营养标记的培养基中生长。因此,当有相互作用的蛋白存在时,激活报告基因的表达,从而能够在不含营养标记的培养基中生长,以此验证是否存在相互作用。
酵母双杂交系统由三个部分组成:
目前酵母双杂交实验采用的系统有LexA系统和Gal4系统两种。在LexA系统中,DNA结合结构域由一个完整的原核蛋白LexA构成,转录激活结构域则由一个88个氨基酸的酸性的大肠杆菌多肽B42构成,它在酵母中可以活化基因的转录;在Gal4系统中,BD和AD分别由Gal4蛋白上两个不同的结构域(1-147aa与768-881aa)构成。

酵母作为酵母双杂交系统的报道株,它具有诸多优点:易于转化、回收扩增质粒;具有可直接进行选择的标记基因和特征性报道基因;酵母内源性蛋白不易同来源于哺乳动物的蛋白结合。
利用酵母双杂交系统可以直接、快速的研究蛋白-蛋白之间的相互作用,具有敏感性高、操作简便等优点
| 酵母双杂交系统 | 免疫共沉淀(Co-IP) | |
|---|---|---|
| 检测瞬时相互作用 | 是 | 否 |
| 检测弱相互作用 | 是 | 否 |
| 验证方式 | 体内相互作用检测 | 体内相互作用检测 |
| 是否需要蛋白纯化 | 否 | 是 |
| 是否需要抗体 | 否 | 是 |
表1:酵母双杂交与免疫共沉淀对比
假阳性是酵母双杂交实验常遇到的问题,假阳性是指两待测蛋白没有相互作用但依然显示出了阳性结果。产生假阳性的原因很多,如某些蛋白质本身具有激活转录功能或在酵母内表达时发挥转录激活作用,使DNA结合结构域杂交蛋白(BD-bait)在无特异激活结构域的情况下可激活转录。另外有些蛋白质表面有对其它蛋白质的低亲和力区,容易形成蛋白体复合物,能够引起报告基因表达,产生假阳性结果。
为了降低假阳性需要设计正确的对照,应对诱饵蛋白和靶蛋白能否单独激活报告基因做验证。使用两个或两个以上的报告基因也可以降低假阳性,利用两个以上报告基因,只有同时具有两种以上相应表型才是真阳性结果。另外, 将报告基因整合到染色体上,可以使基因表达水平稳定,消除了由于质粒拷贝数变化引起基因表达水平波动而造成的假阳性。
酵母双杂交分析相互作用的蛋白必须定于核内才能激活报告基因,但是很多蛋白质的相互作用依赖于翻译后加工,如二硫键的形成等,而此过程需要在包浆内完成,这样有多种蛋白质不适用此方法。
为了解决这一问题,人们对酵母双杂交技术进行改进,发展了核外双杂交技术(见下文)。
在传统酵母双杂交系统的基础上,发展了多种改型的双杂交技术以适应不同目的需要。
反向杂交技术(也成反式双杂交技术)主要用于在确定蛋白质之间相互作用后,进一步研究其结构和功能的关系,确定蛋白质间作用的关键位点或起决定作用的个别氨基酸、特定结构等。这项技术的特点是采取了反选择筛选策略,其关键是报道基因的表达产物对细胞生长有抑制作用。这样当诱饵蛋白与靶蛋白存在相互作用时,表达出有毒性的报道蛋白,细胞不能生长。
例如Vidal等采用了反式选择性的报告基因URA3,其编码的酶使细胞不能在5一氟乳清酸存在的情况下下生长。如果融合蛋白的相互作用被阻断, URA3则不表达,酵母菌表现对5一氟乳清酸的抗性。因此,采用反式双杂交系统可以很容易地检验出与某一蛋白相互作用的蛋白的突变体。对突变体进行研究,即可找出影响蛋白质间相互作用的关键位点。

酵母三杂交系统可以用于检测蛋白质-核酸之间的相互作用。三杂交系统的本质与双杂交是相同的,只是需通过第三个分子的介导把两个杂交蛋白带到一起。它的基本原理是将一个已知与RNA结合的蛋白与BD构建第一个融合蛋白,将待测蛋白质与AD构建第二个融合蛋白,同时构建一条含有两个不同结合位点的融合RNA,当RNA与两个融合蛋白相结合时,可激活报道基因的表达。除了蛋白质-核酸之间的相互作用,酵母三杂交系统也可用于检测检测蛋白质与肽配体、蛋白质与有机小配体、蛋白质与蛋白激酶间的相互作用。
在传统的酵母双杂交系统中,蛋白之间的相互作用是在细胞核内发生的。因此,它不能检测某些在核外蛋白之间的相互作用。为了克服这种局限性,发展出了核外双杂交技术。该技术以SRS 和 USPS两种系统为代表。
SRS 也称 Sos 蛋白召集系统 (Sos RecruitmentSystem) 。它的基本原理是分别将待测蛋白X与哺乳动物细胞的一种鸟苷交换因子( EGF) Sos蛋白融合;将Y蛋白与锚定在酵母细胞膜上的Src肉豆寇烯化信号蛋白融合 ,并使它们共表达于一个cdc25-2基因温度敏感型突变的酵母菌株内。由于该菌株中cdc25-2基因编码的 EGF 蛋白在 36 ℃条件下不能激活细胞膜上的Ras蛋白,Ras途径不通。所以细胞无法在36 ℃条件下生存。但如果待测蛋白X与Y之间发生相互作用就能把Sos蛋白带到细胞膜上并激活附近的Ras蛋白,从而打通Ras途径,使该菌株获得在36℃条件下生存的能力。
USPS也称为基于遍在蛋白的裂解蛋白感受器。它的设计是根据这样一个事实,即真核细胞中遍在蛋白与某一蛋白之间新生成的融合会被遍在蛋白特异的蛋白酶(UBPs) 迅速切开,但这种切割只有当遍在蛋白正确折叠时才会发生。研究发现,遍在蛋白基因的N端和C端这两部分即使分离,只要共表达与同一个细胞内,它们仍能正确折叠。将待测蛋白X与带有点突变的遍在蛋白N端片段融合,将待测蛋白Y与下游接有报告蛋白的正常遍在蛋白C端片段融合,并使它们共表达与同一个细胞内。因遍在蛋白N端片段内含有点突变,它与C端片段之间不能自然形成正确的折叠。只有当蛋白X和Y之间发生相互作用时才能克服点突变的影响,使遍在蛋白的两端形成正确折叠,从而引来UBPs切除与C端片段连接的报告蛋白。此技术建立之初是通过Western blot检测有无被切下的报告蛋白来判断待测蛋白之间是否发生了相互作用的,所以操作比较繁琐,不便于推广。最近有人对它作了改进 ,以一种融合的转录激活因子PLV作为报告蛋白。PLV一旦被从遍在蛋白C端片段上切下,它就会进入细胞核内激活特定的报告基因 ,如LacZ和HIS3等。这样就可以根据转化细胞是否生长及显色来判断待测蛋白之间有无相互作用。因此极大地简化了操作步骤,提高了筛选效率。
Fret荧光共振能量转移检测蛋白质间相互作用
TAP串联亲和纯化技术检测蛋白质间相互作用
答:input是阳性对照。免疫共沉淀实验中,会直接取细胞裂解液进行WB,用于验证细胞裂解液中确实存在目的蛋白,即阳性对照。
答:最好选择两个不同种源的抗体。选择同一种源的抗体的问题在于:在WB之前进行的蛋白变性这一步,由于加样缓冲液中含有巯基乙醇,巯基乙醇会把抗体的重链和轻链之间的二硫键破坏,从而使的抗体变成重链分子55KD和轻链分子25KD。假设你IP的抗体是兔抗的,目的蛋白进行WB的抗体也是兔抗的,而且目的蛋白的分子量为55KD,可以想象,55KD的位置除了你的目的蛋白以外,还有IP抗体的重链IgG。由于IP的抗体用量非常大(1ug),就导致55KD处的信号会非常强。如果你用抗兔的二抗进行显色,那么55KD处的ip抗体的重链和你的目的蛋白会重叠在一起,无法分辨。
然而这个时候如果你的目的蛋白进行WB的抗体不是兔抗的,比如小鼠抗的,那么就需要用抗小鼠的二抗进行显色,而抗小鼠的二抗是无法跟兔抗的IP抗体发生反应的。因此55KD位置的IP抗体的重链IgG就无法显色,发生反应的就是你的目的蛋白了。
答:若抗体浓度高,则降低抗体浓度;若抗体特异性不好,则换抗体;若PCR污染,则重新配置缓冲溶液。
答:ProteinA/G能特异性地结合到免疫球蛋白的FC片段,因此能和抗体结合,而抗体与目标蛋白结合,目标蛋白和相互作用的蛋白结合。
答:排出污染的可能性,例如检测一个兔子细胞的某种蛋白质,若没有设置LgG的对照,而操作中有非特异性蛋白质,又和设计的抗体有作用,就会出现阳性的结果。如果做了阴性对照,也就是lgG对照,对照没有出现阳性就说明没有非特异性的蛋白,对照出现阳性,说明抗体有非特异结合的可能,需要重新开始实验。
答:用兔的lgG做对照;用抗Fab和Fc抗体特异性封闭轻链和重链;在WB二抗上着手,这种二抗针对的是轻重链间的二硫键,若跑SDS胶,最后显影是检测不到重链和轻链的。
答:裂解液中的去垢剂浓度太高或配方过于剧烈:此时降低去垢剂浓度或更换去垢剂种类;
受蛋白的亚细胞定位影响:应重新选择裂解液配方来释放目的蛋白;
蛋白与蛋白之间的相互作用太弱或不太稳定:应选择亲和力更高的抗体以捕获更多的目的蛋白,从而捕获更多的相互作用蛋白;过表达提高目的蛋白的含量:选择目的蛋白或相互作用蛋白含量高的样品进行免疫共沉淀实验。
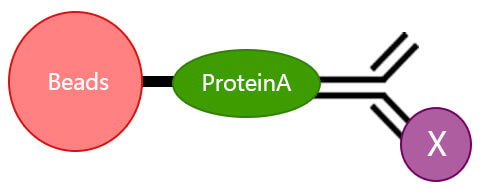 免疫沉淀
免疫沉淀免疫沉淀(immunoprecipitation)是指利用抗体可与抗原特异性结合的特性,将抗原(常为靶蛋白)从混合体系沉淀下来,初步分离靶蛋白的一种方法。
免疫共沉淀(coimmunoprecipitation)是一种在体外探测两个蛋白分子间是否存在特异性相互作用的一种方法。其原理非常简单,如果两个蛋白在体外体系能够发生特异性相互作用的话,那么当用一种蛋白的抗体进行免疫沉淀时,另一个蛋白也会被同时沉淀下来。与酵母双杂交技术不同,免疫共沉淀技术所利用的是抗原和抗体间的免疫反应,是一种基于体外非细胞的环境中研究蛋白质与蛋白质的相互作用的方法。
免疫共沉淀与免疫沉淀技术所使用的原理与方法大致相似,所不同的是,在免疫共沉淀中,对靶蛋白的结合与沉淀由另一个与之发生相互作用的蛋白替代。在免疫共沉淀或免疫沉淀的基础上,通过进一步与其它技术的结合,如聚丙烯酰胺凝胶电泳,还可进一步对靶蛋白的的分子量等特性进行鉴定。
答:非特异蛋白结合,处理方法:在无血清培养液中裂解细胞; 在免疫沉淀前用protein (G/A)珠子预洗免疫沉淀后增加漂洗次数和严谨度(高盐或去垢剂),裂解液严谨度太低,处理方法:改用高严谨度裂解液,实验仪器或液体被污染,处理方法:使用洁净的仪器或液体转移膜上的非特异吸附,处理方法:戴手套, 用镊子夹取, 不要接触膜转移面。
样品被蛋白酶降解,处理方法:添加蛋白酶抑制剂(protease inhibitor);所有操作保持4℃以下冰上操作并防止冻融。
抗体浓度太低,处理方法:调整IP和/或IB抗体浓度, 必要时设立浓度梯度,摸索最佳浓度;
抗体亲合力太低,处理方法:选用适合于IP和/或IB的相应抗体;
IP抗体未与agarose珠子结合,处理方法:选用适合于IP的相应珠子, 正确保存防止变质或干燥;
Tag未暴露在融合蛋白构象的表面,处理方法:改变tag融合表达部位;
裂解液严谨度太高,处理方法:改用低严谨度裂解液。
答:一般有三种反应顺序:1.样本+抗体先反应,然后加磁珠;2.抗体+磁珠先反应,然后放入样本中;3.样本+抗体+磁珠同时反应。推荐使用第一种和第二种,差别不大。不推荐用第三种,第三种方式虽然可以减少反应时间,但有实验表明同时加入三个组分的方式会使最终的结果变差。
对于分子生物学来讲,生物分析手段的发展,是阐明机理的必要条件。在研究分子间相互作用的道路上,人们不断探索,总结出很多方法,免疫技术,晶体衍射,核磁共振等。1948年,荧光共振能量转移(Fluorescence resonance energy transfer,FRET)理论被首次提出,它可以测定1.0-6.0nm距离内分子间的相互作用。1967年,这一理论得到了实验验证,将1.0-6.0nm的距离称为光学尺。二十世纪八十年代出,通过科学家的不断探索,Fret技术成功运用到蛋白质结构的研究中。自Fret荧光共振能量技术诞生以来,已结合多种先进的技术和方法,如电子显微镜,X射线衍射等,推动了分子生物学检测手段的发展。
荧光共振能量转移技术,是采用物理方法去检测分子间的相互作用的方法。他适用于在细胞正常的生理条件下,验证已知分子间是否存在相互作用。此方法的检测原理如下;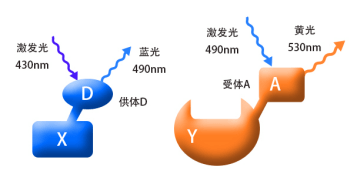
将我们要检测的蛋白(如图X和Y),分别偶联上D和A荧光蛋白,D和A是一对荧光物质,我们称之为供体(donor)和受体(acceptor)。当用430nm的紫光去激发X融合蛋白时,它能够产生490nm的蓝色荧光;同样,当我们用490nm的蓝光去激发Y融合蛋白时,它能够产生530nm的黄色荧光。(结合图1) 。
当蛋白X和Y间没有相互作用时(两者的空间距离>10nm),融合蛋白X和Y分别产生相应的荧光而被检测到,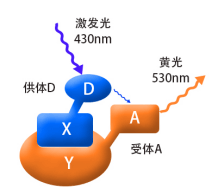 如果蛋白X和Y间存在相互作用(两者的空间距离需<10nm,结合图2),用紫光激发融合蛋白X其产生的蓝光会被融合蛋白Y吸收,从而产生黄色荧光,这时,在细胞内将检测不到蓝色荧光的存在。这时因为能量从X融合蛋白转移到了Y融合蛋白,这就是荧光共振能量转移技术。
如果蛋白X和Y间存在相互作用(两者的空间距离需<10nm,结合图2),用紫光激发融合蛋白X其产生的蓝光会被融合蛋白Y吸收,从而产生黄色荧光,这时,在细胞内将检测不到蓝色荧光的存在。这时因为能量从X融合蛋白转移到了Y融合蛋白,这就是荧光共振能量转移技术。
一个理想的Fret相互作用体系,要求要有一对合适的荧光物质, 即供体的发射光谱与受体的吸收光谱有明显的重叠。且当供体的激发波长时对受体无影响,供体和受体的发射光谱要完全分开,否则容易造成光谱干涉,而使反应体系不稳定。目前,较为常用的供体-受体分子对,主要有绿色荧光蛋白类(GFPs)和染料类。绿色荧光蛋白类有CFP-YFP,BFP-GFP,BFP-YFP等,染料类的有Cy3-Cy5,FITC-Rhodamine等。且这些荧光物质要能够标记在研究对象上。
| 优点 | 缺点 |
|---|---|
| 在活细胞的正常生理条件下进行检测,观察大分子在细胞内的构象变化与相互作用,并弥补了需破碎细胞检测相互作用的缺点; 灵敏度高,可实现对单细胞水平的研究,研究单个受体分子; 可与多种仪器和技术结合使用,如显微镜,色谱技术,电泳,流失细胞技术等; |
应用比较局限,一般需要在待检测分子上偶联荧光物质(加上标记); 对实验要求较高,如供受体的光谱重叠不好,会导致荧光干扰,对供受体的抗干扰能力,水溶性等要求高; 需要不断探索合适的供体和受体,且能够标记分子; 难以观察瞬时的分子间作用,检测要求大量的样品; |
以荧光物质CFP(供体)-YFP(受体)为例,检测AB蛋白在细胞内的相互作用。
相关服务:Co-IP蛋白互作检测服务
免疫共沉淀(Co-IP)是利用抗原与抗体之间的专一性作用为基础,用于研究蛋白质与蛋白质之间相互作用的经典方法。利用免疫共沉淀可以检测两个已知蛋白之间的相互作用,或者利用已知蛋白寻找与之相互作用的未知蛋白。相较于其他分子间相互作用检测方法(GST pull down等),免疫共沉淀实验的优势在于蛋白的结合在细胞内完成,能够反应天然状态下的蛋白质相互作用,结果更加真实可靠。

当细胞在非变性条件下被裂解时,完整细胞内存在的许多蛋白质-蛋白质相互作用被保留了下来,假如细胞内存在XY蛋白复合物,用X(X也称为诱饵蛋白)的抗体免疫沉淀X,那么与X在体内结合的蛋白质Y(Y也称为靶蛋白)也被沉淀下来。因此在细胞裂解液中加入X的抗体沉淀蛋白X,随后利用蛋白印迹(western blot,WB)检测沉淀中是否存在蛋白Y,如果存在,则说明细胞内存在XY蛋白复合物,即蛋白XY存在相互作用。
免疫共沉淀实验流程为:收集蛋白样品—诱饵蛋白抗体沉淀诱饵蛋白—SDS-PAGE分离蛋白—WB检测是否存在靶蛋白。详细实验操作流程请至“免疫共沉淀实验操作流程及注意事项”查看。
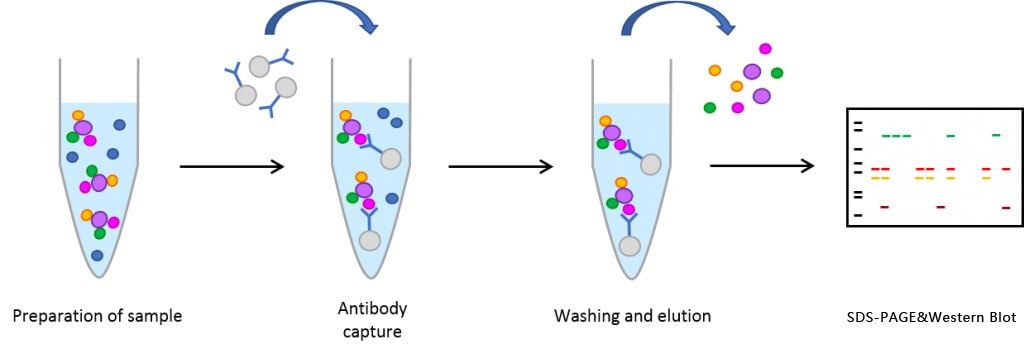
蛋白样品通常通过裂解细胞得到。Co-IP实验通常有两种,一种为内源性蛋白相互作用验证,另外一种为非内源性蛋白相互作用验证,内源性相互作用是指两蛋白相互作用在细胞内发生,即通过质粒共转染的方式将两种蛋白转染至同一细胞内表达,表达完成后收集细胞进行裂解得到蛋白样品。非内源性相互作用两蛋白相互作用在细胞外发生,即将含有靶蛋白的动植物组织、器官进行预处理及细胞裂解获得细胞裂解液,随后将诱饵蛋白(通常为重组表达纯化获得)加入细胞裂解液中,得到蛋白样品。
利用磁珠偶联抗体沉淀诱饵蛋白:在样品中加入磁珠偶联抗体,抗体会与诱饵蛋白结合,利用磁铁将磁珠拉下,同时诱饵蛋白会一起被沉淀出来。如果存在与诱饵蛋白的相互作用的靶蛋白,也会被沉淀下来。如果没有诱饵蛋白的特异性抗体,可以给诱饵蛋白加上标签(Myc、HA、Flag等),然后利用标签抗体沉淀蛋白。
得到沉淀后,需要验证沉淀中是否存在相互作用蛋白。先利用SDS-PAGE将蛋白复合物分离,随后利用WB检测是否存在靶蛋白(如果靶蛋白的分子量是已知的,可以直接用SDS-PAGE检测是否存在靶蛋白)。
为了确保最终得到的结果是天然状态下的相互作用,而不是由于某些方面造成的人工相互作用(也就是“假阳性”),在Co-IP的实验设计过程中,需要设置正确的对照。
假设Co-IP实验的实验组为“磁珠+抗X抗体+靶蛋白X+目的蛋白Y”,则可能出现的“假阳性”及对应需要设置的实验对照如下表所示:
| 可能出现的“假阳性” | 需要设置的对照实验 |
|---|---|
| 磁珠与蛋白X非特异性结合 | “磁珠+抗体X+抗体Y” |
| 磁珠与蛋白Y非特异性结合 | “磁珠+蛋白Y” |
| 抗X抗体和Y非特异性结合 | “磁珠+抗体X+蛋白Y” |
| 抗X抗体(或磁珠)会和细胞裂解液内其它蛋白结合 | “磁珠+抗体X+未转染质粒的宿主细胞裂解液” |
| 抗体的非特异性结合 | “normal IgG+蛋白X+蛋白Y” |
上述为了避免出现“假阳性”的对照称为阴性对照,除此之外Co-IP实验还会设置一个阳性对照组(Input组),Input组为直接利用抗体X(抗体Y)对细胞裂解液进行WB检测,验证细胞裂解液中存在蛋白X(抗体Y)。
在某些Co-IP实验中,实验人员会把IP后的上清分别进行蛋白X和蛋白Y的WB检测,该对照组称为output组。
利用内源性Co-IP实验为验证两个蛋白是否存在已知作用,如果最终的结果为阳性,则可以证明两个蛋白之间存在相互作用;但如果结果为阴性,无法证明两个蛋白之间不存在相互作用,也有可能是蛋白在细胞内表达量低等原因导致。因此在进行内源性Co-IP验证两个蛋白是否存在相互作用时,建议先做过表达Co-IP作为对照。
在免疫共沉淀实验中要保证实验结果的真实性,应注意以下几点:
免疫共沉淀实验可以用于:
随着对蛋白质研究的不断深入,人们将免疫沉淀方法与其他方法结合起来,在其基础上衍生出许多较为复杂的技术,从而使得分析方法更为多样化,它的应用范围也相当的广泛。免疫共沉淀是用来研究蛋白质与蛋白质相互作用的一种技术,可以应用于蛋白复合物的研究。它可验证蛋白复合物的存在,进而发现新的蛋白复合物;免疫共沉淀技术与免疫印迹法或质谱等方法结合,用于确定诱饵蛋白-目的蛋白在天然状态下的结合情况,确定特定蛋白质的新作用搭档。免疫共沉淀实验也可以应用于低丰度蛋白的富集和浓缩。
同时免疫共沉淀技术是一个相对比较经典的探讨蛋白质间相互作用的技术,在现代医学研究中应用范围广泛且可信度较高。蛋白质之间相互作用渗透于机体每一个细胞的生命活动过程中,生物学中会出现很多现象如复制、转录、翻译、剪切、分泌、细胞周期调控、信号传导和中间代谢等都受到蛋白质间相互作用的调控。有些蛋白质由多个亚单位组成,蛋白质之间的相互作用就显得尤为普遍。又有些蛋白质结合得十分紧密;而有些蛋白质却只有短暂的相互作用。可是不论出现哪些种情况,它们都控制着大量的细胞生命活动的事件,比如细胞的增殖、分化和死亡。且通过蛋白质之间的相互作用,能改变细胞内蛋白质的动力学特征,比如底物结合特性、催化活性,也可以产生新的结合位点,对改变蛋白质对底物的特异性有作用,还可以使其他蛋白质失活,其他基因表达得到调控。因此,只有让蛋白质之间相互作用得以顺利进行,细胞的正常生命活动过程才会得到保障。因为蛋白质之间相互作用具有如此重大的意义,所以它的检测方法的研究也备受关注与重视。自此以后蛋白相互关系的研究会愈演愈烈,未来不仅仅可以通过免疫共沉淀技术来证实,还将会有越来越多的先进技术值得去应用和发展。
本文主要介绍了GST pull-down实验的服务流程(数据以实际实验为准)
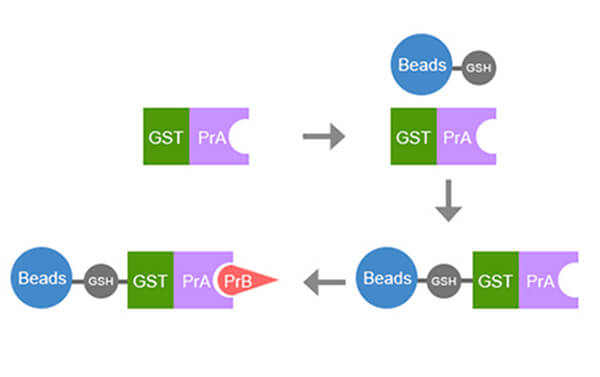
gst pull down 实验流程
相关服务:GST pull-down服务
相关服务:免疫共沉淀检测(CO-IP)
免疫共沉淀(Co-Immunoprecipitation)是以抗体和抗原之间的专一性作用为基础的用于研究蛋白质相互作用的经典方法。是确定两种蛋白质在完整细胞内生理性相互作用的有效方法。 免疫共沉淀实验的操作步骤比较多,要得到一个完美的实验结果,不仅需要高质量的抗体,对免疫沉淀体系也需要有严格的控制指标。在免疫共沉淀的实验过程中,从蛋白样品处理、抗体-琼脂糖珠复合物洗涤到最后的鉴定,每步都非常关键,需要严格控制实验流程中每个关键步骤的质量,才能达到最终的实验目的。
该免疫共沉淀操作步骤是将抗体先结合到蛋白A/G琼脂糖珠上,然后再与抗原混合。相对其他方法,最终的得率较低,但避免了抗体共洗脱的问题。如果希望获得高纯度的目的蛋白,而不考虑非特异性结合,可以将抗体和蛋白样品在加入蛋白A/G琼脂糖珠之前进行混合,这样最后抗体也会和目的蛋白一同被洗脱下来,从而可能会对蛋白质印迹法(western blot)检测造成干扰。
注:RIPA配制:150mM Nacl,1%乙基苯基聚乙二醇,1%脱氧胆酸钠,25mM Tris-HCl缓冲液(PH7.6),0.1%SDS。
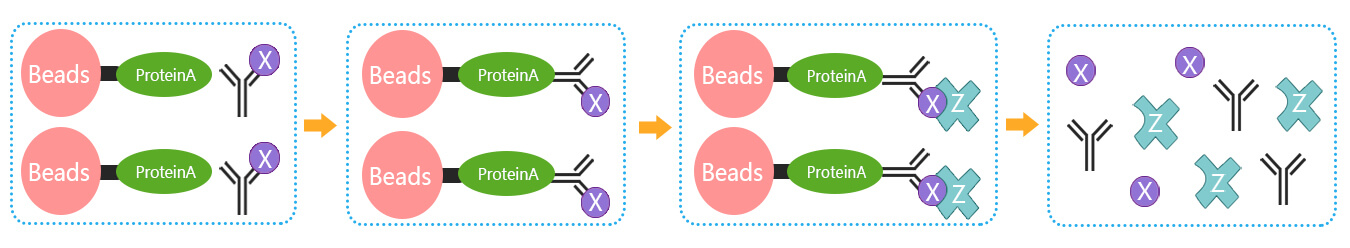
免疫共沉淀实验流程
该实验的思路为:先通过免疫共沉淀实验,得到结合蛋白,随后对得到的结合蛋白进行测序。
1)细胞裂解采用温和的裂解条件,不能破坏细胞内存在的所有蛋白质-蛋白质相互作用,多采用非离子变性剂(NP-40 或Triton- X-100)。每种细胞的裂解条件是不一样的,通过经验确定;
2)不能用高浓度的变性剂(0.2%SDS),细胞裂解液中要加各种酶抑制剂;
3)使用对照抗体:单克隆抗体:正常小鼠的IgG或另一类单抗;兔多克隆抗体:正常兔IgG;
4)确保共沉淀的蛋白是由所加入的抗体沉淀得到的,而并非外源非特异蛋白,单克隆抗体的使用有助于避免污染的发生;
5)要确保抗体的特异性,即在不表达抗原的细胞溶解物中添加抗体后不会引起共沉淀;
6)确定蛋白间的相互作用是发生在细胞中,而不是由于细胞的溶解才发生的,这需要进行蛋白质的定位来确定。
不同反应的“强”“弱”的标准:
动力学是通过实时的结合解离曲线计算得到解离常数和结合常数来更细致的描述相互作用。一般只能通过实时监测的技术获得, 比如 BLI。亲和力是描述反应强弱, 基于终点技术的很多方法可以获得, 比如 ELISA。动力学包含了亲和力, 但是亲和力不包含动力学。
如果需要获得 KD 值,必须要知道分析物的浓度。KD 值越大,需要的分析物的测试浓度越大。
小到150 Da的化合物,大到病毒,以及多肽,蛋白质,DNA/RNA,多糖,多聚物,脂质体,纳米颗粒等均在
BLI的可测试范围。
如果是 SA,SAX,SSA,AR2G,APS 传感器,固化蛋白一定要纯化。如果是基于capture类传感器,固化蛋白不一定需要纯化。但是固化纯化的物质可以减轻杂质带来的影响。
信号和结合上去的分子的大小和密度有关。对不同物质来说,分子越大,信号越高。对同一种物质来说,结合越多,信号越高,结合量和信号呈正比。
小到150 Da的化合物,大到病毒,以及多肽,蛋白质,DNA/RNA,多糖,多聚物,脂质体,纳米颗粒等均在BLI的可测试范围。
生物素试剂与蛋白的摩尔比在 1:1 – 3:1 之间。目前使用最多的是氨基偶联的生物素化试剂,所以待生物素化的物质溶液中除了需要生物素化的物质外,不得含有其他带有氨基的保护剂以及缓冲液 (特别是 Tris 缓冲液)。生物素化完成后需要脱盐除去未反应的生物素化试剂。






南京德泰生物工程有限公司 Nanjing Detai Bioengineering Co.,Ltd. ©2025 All Rights Reserved
 苏公网安备32011202001300
苏公网安备32011202001300