His标签亲和纯化实验步骤
His标签是目前蛋白表达中最常用的标签,而且很成熟,它的优点是表达方便而且基本不影响蛋白的活性,无论是表达的蛋白是可溶性的或者包涵体都可以用固定金属离子亲和色谱去(IMAC)进行蛋白纯化。
Ni柱的选择
镍离子有六个螯合价位,Ni-NTA螯合了四价,剩余两价;而Ni-IDA螯合三价,剩余三价。因此Ni-IDA结合作用力要比Ni-NTA 强,在同样条件下Ni-IDA 的载量要比Ni-NTA高,而且洗脱杂质和目标蛋白所用的缓冲液中咪唑浓度更高;但Ni-NTA更稳定,耐受更强的还原剂,镍离子更不易脱落。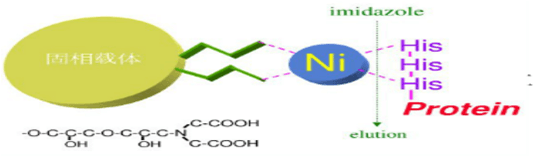
破碎方法
样品的处理对纯化是很重要的,重要的原则是破碎要温和,不能使蛋白断裂或者降解,否则一些片段同样也带有标签,这样增加了纯化的难度。需要注意的问题是超声破碎温度,强度,时间。
His标签亲和纯化
可溶性蛋白的破碎
- 收集培养发酵液,20℃,5000rpm离心8分钟,沉淀转移到离心管中,10℃,13000rpm离心1分钟,收集沉淀的菌体(如果不是马上破碎可以放-20度冷冻。)
- 取8-10克菌体加40-50ml破碎缓冲液(例:20mM Tris,pH8.0,0.15M NaCl,1%Triton X-100,pH8.0)。加入Leupeptin与Pepstantin终浓度1ug/ml,加入TCEP终浓度1mM/ml。
- 把混合菌体在冰水中用超声探头破碎,超声3S,间隔8S。超声15-20Min。(超声完全的判断:溶液中有无块状物,溶液的黏稠度)
- 破碎的液4℃13000rpm离心10分钟,取上清,再次4℃13000rpm离心10分钟,然后把上清和沉淀分别留样,先做上清纯化,如果只沉淀中有目标蛋白,那就用变性条件下去提取。
- 取纯化Ni柱,清洗干净柱子,加入3-4ml镍琼脂糖凝胶,用洗瓶清洗柱子5次,再用平衡液平衡柱子2次(PS:平衡液即破碎液,但不含有Triton X-100。)
- 将柱子和上清充分结合,放置四维旋转混匀器上孵育1-2h后拿出纯化
可溶性蛋白的纯化
- 平衡缓冲液:pH8.0的50mM Tris缓冲液含0.15M NaCl,含20mM咪唑。
- 洗脱缓冲液:① pH8.0的50mM Tris缓冲液含0.15M NaCl,含50mM咪唑。
② pH8.0的50mM Tris缓冲液含0.15M NaCl,含100mM咪唑。
③ pH8.0的50mM Tris缓冲液含0.15M NaCl,含300mM咪唑。
- 过柱,流出FT,取样FT。
- 用20倍柱体积平衡缓冲液洗去未吸附的样品,流速1-2ml/min,直到流出液点样G250显示无蓝色是开始洗脱柱子上吸附的蛋白。
- 用50mM,100mM,300mM咪唑浓度分阶段洗脱柱子,洗脱5-10个柱体积左右(PS:蛋白吸附较多,则用多的洗脱液洗脱。),流速1-2ml/min,4ml/管收集。(收集蛋白起点的确定:紫外分光光度的示数上升较快、考马斯亮蓝G250点样确定变蓝色;收集蛋白终点的确定:考马斯亮蓝G250点样无明显蓝色,紫外分光光度示数下降平稳。)
- 将收集的各浓度梯度洗脱下来的蛋白,取样,跑SDS-Page。
- 根据SDS-Page电泳图,若有蛋白,则收集蛋白进行透析,若电泳结果无蛋白,而沉淀中有蛋白,则用变性条件下洗沉淀,做沉淀纯化。
包涵体破碎
1、大肠杆菌的破碎方法:
1) 收集培养发酵液,20℃,5000rpm离心8分钟,沉淀转移到离心管中,13000rpm,10℃,收集沉淀的菌体(如果不是马上破碎可以放-20度冷冻。)
2) 取5-10克菌体加40-50ml破碎缓冲液(50mM Tris, pH8.0,0.15M NaCl,1%Triton X-100,pH8.0)加入Leupeptin与Pepstantin终浓度1ug/ml,加入TCEP终浓度1mM/ml。
3) 把混合菌体在冰水中用超声探头破碎,超声3S,间隔8S。超声15-20Min。(超声完全的判断:溶液中有无块状物,溶液的黏稠度)
4) 破碎的液4℃13000rpm离心20分钟,然后把上清和沉淀分别留样,取沉淀。
5) 沉淀加入再次破碎缓冲液( pH8.0,50mM Tris,2mM EDTA,150mM NaCl,20mM咪唑,1% Triton X-100。)把混合菌体在冰水中用超声探头破碎,超声3S,间隔8S。超声15-20Min。
6) 破碎的液4℃13000rpm离心20分钟,取沉淀。(PS:沉淀中不能含有溶液,所以必须要倒干净。)
7) 破碎离心的沉淀先加缓冲液(20mM咪唑,20mMTris,pH8.0)1ml溶解沉淀,加变性溶液( pH8.0的50mMTris缓冲液含0.15 NaCl,8M脲)。(加入溶液比例,1g沉淀加入溶液4-5ml。)
8) 把混合菌体在冰水中用超声探头破碎,超声3S,间隔8S。超声10Min。
9) 破碎的液4℃13000rpm离心10分钟,取上清,再次4度13000rpm离心10分钟,取上清。
10) 在做可溶性蛋白纯化时,沉淀中有,但是上清没有的情况下,取沉淀,直接从(5)步骤往下操作。
包涵体蛋白的纯化
- 平衡缓冲液:pH8.0的50mM Tris缓冲液含0.05M NaCl,8M脲。
- 洗脱缓冲液:①pH8.0的50mM Tris缓冲液含0.05M NaCl,8M脲,含20mM咪唑
②pH8.0的50mM Tris缓冲液含0.05M NaCl,8M脲,含50mM咪唑
③pH8.0的50mM Tris缓冲液含0.05M NaCl,8M脲,含100mM咪唑
④pH8.0的50mM Tris缓冲液含0.05M NaCl,8M脲,含300mM咪唑
⑤pH8.0的50mM Tris缓冲液含0.05M NaCl,8M脲,含500mM咪唑。
- 取3-4ml镍琼脂糖凝胶FF,用洗瓶清水洗清柱子5次,用50ml平衡缓冲液平衡。
- 将柱子和上清充分结合,放置四维旋转混匀器上孵育1-2h后拿出纯化。
- 过柱,流出FT。
- 用20倍柱体积平衡缓冲液洗去未吸附的样品,流速1-2ml/min,直到流出液,点样G250显示无蓝色是开始洗脱柱子上吸附的蛋白。
- 用20mM,50mM,100mM,300mM咪唑浓度分阶段洗脱柱子,洗脱5-10个柱体积左右(PS:蛋白吸附较多,则用多的洗脱液洗脱。),流速1-2ml/min,4ml/管收集。(收集蛋白起点的确定:紫外分光光度的示数上升较快、考马斯亮蓝G250点样确定变蓝色;收集蛋白终点的确定:考马斯亮蓝G250点样无明显蓝色,紫外分光光度示数下降平稳。)
- 将收集的各浓度梯度洗脱下来的蛋白,取样,跑SDS-Page。
- 根据SDS-Page电泳图获取目标蛋白所在咪唑浓度梯度,收集蛋白,透析获的目标蛋白
常见问题及解决方案
- 蛋白不溶解或沉淀在柱子上:要留意缓冲液体的pH,根据蛋白的PI值确定缓冲液和洗脱液的pH值,通过增加盐浓度,避免蛋白的非特异性吸附,蛋白洗脱时在柱中沉淀 ,变性条件下进行洗脱。以在离心管中结合、洗脱的小量批次方式进行纯化,以避免局部蛋白浓度过高。
- 蛋白不吸附:这是最常见的,通常的原因有①是标签不暴露,被折叠在蛋白的结构内,可以在变性的条件下去纯化,②可以选择作用力更强,配基密度更高的填料,通常镍琼脂糖凝胶作用力最强,如果蛋白分子量大可以选择手臂长的填料,如镍NTA琼脂糖凝胶,填料的好坏可以看填料的颜色,颜色越深那配基密度越高,作用力也相应要强。③样品的pH过低或者沉淀导致不能吸附,所以样品和缓冲液的pH要尽量一致,避免沉淀。④加吐温可以可以降低表面张力,同时也可以降低疏水相互作用力了,使蛋白不会因为别的作用力而被吸附到柱子上,同时增强了洗脱能力。⑤ 缓冲液条件不正确,检查漂洗缓冲液的pH值和组成,确保体系中不含鳌合剂及还原剂。
- 难洗脱:如果穿透中目标蛋白明显减少,而洗脱又没有,取点填料加电泳缓冲液煮后离心跑电泳还是有目标蛋白,可以用更强的洗脱条件如500mM咪唑,如果还不能洗脱,可以直接用500mM咪唑加8M脲去洗脱。
- 目的蛋白与杂蛋白一起洗脱出来:①色谱柱载量相对过大,减少Ni-NTA树脂或Ni-IDA树脂用量;②杂蛋白与目的蛋白结合,共纯化下来,加大漂洗缓冲液的盐浓度,或增加去污剂浓度,加入乙醇/甘油以破坏蛋白之间的疏水作用。
相关服务







 苏公网安备32011202001300
苏公网安备32011202001300