
自德国诺贝尔奖获得者保罗·埃利希(Paul Ehrlich)最早于1913年提出ADC药物的概念至今,ADC药物经历了三代创新。ADC药物进入血液循环并与肿瘤细胞表面的靶抗原受体结合后,新形成的ADC抗原复合物在内化后会被溶酶体降解,释放有效载荷并诱导肿瘤细胞死亡(图1)。有效载荷是ADC设计的重要组成部分,其作用机制是决定ADC性能的重要因素。
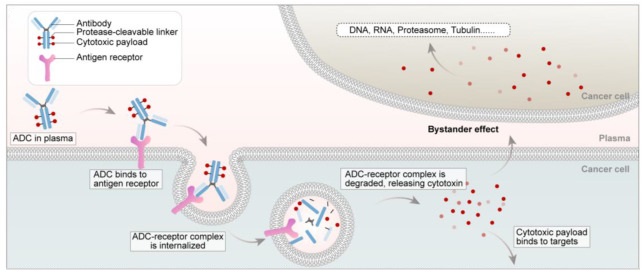
图1 ADC药物作用机制
第一代ADC药物多选用甲氨蝶呤、长春碱和多柔比星等传统化疗药物作为细胞毒性有效载荷。然而,由于它们对癌细胞的细胞毒性不足,在靶细胞中的低积累,疗效不及亲本有效载荷,导致研发在临床阶段失败。随后,第二代ADC药物的有效载荷聚焦在新型高细胞毒性化合物,其活性比第一代ADC中使用的传统化疗药物强100至1000倍。当它们单独使用时,往往伴随难以接受的严重副作用,因此多未能作为用于癌症治疗单一药物被批准。有趣的是,这些高细胞毒性化合物是ADC药物理想的有效载荷。大多数第二代ADC有效载荷为微管蛋白抑制剂,虽然对活跃分裂的肿瘤细胞非常有效,但对静态癌细胞的效果要差得多。为了克服这一局限性,第三代ADC药物多选择可以靶向整个细胞周期的DNA损伤剂作为有效载荷。DNA损伤剂可以通过双链断裂、烷基化、嵌入和交联来破坏DNA结构以杀死肿瘤细胞。
有效载荷的活性和理化性质直接影响ADC药物的抗肿瘤疗效,其细胞毒性、免疫原性、制备和循环过程中储存的稳定性、水溶性和可改造性也很重要。理想的有效载荷应具有以下特征:
(1)高细胞毒性。肿瘤特异性抗原非常有限,尤其是在实体瘤中。此外,由于单克隆抗体的通透性低,内化活性差,通过抗体抗原结合可以内吞到肿瘤细胞中的ADC有效载荷数量非常少。
(2)低免疫原性。尽管目前ADC药物的抗体部分使用人源或人源化单抗,但仍有诱导免疫反应的风险,这可能会对ADC的疗效产生负面影响。一些从植物、动物或微生物中提取的剧毒有效载荷,可以确保在人体中的免疫原性足够小,甚至可以忽略不计。使用较小分子量的有效载荷也是降低免疫原性风险的一种方法。
(3)高稳定性。由于抗体在循环中的半衰期很长,因此ADC药物应在血液循环中保持稳定,以避免释放或分解。有效载荷还应在细胞质和溶酶体中保持稳定,在低pH条件下不会发生明显降解。
(4)具有可以改造的官能团。有效载荷必须具有可修饰的官能团或可与单克隆抗体偶联的位点。必须仔细选择修饰的部位,以保持亲本药物的效力。更重要的是,当使用不可切割的接头时,即使在抗体降解后,有效载荷也必须保持效力。
(5)具有旁观者杀伤效果。一些ADC药物被内化后可释放出不带电荷的、易透过细胞膜的小分子,这些小分子通过细胞膜扩散并杀死相邻抗原阴性表达的肿瘤细胞。这一过程被称为“旁观者杀伤效应”,对抗原表达不均匀的肿瘤细胞具有重要意义。一般来说,促进癌细胞中旁观者杀伤效应的有效载荷更适合靶抗原低表达或异质表达的癌症。要构建具有旁观者效应的ADC药物,其结构必须满足连接子可以被裂解,有效载荷可渗透细胞膜的条件。具有强旁观者杀伤作用的有效载荷,也可能容易被健康组织吸收,导致严重的全身毒性,如何平衡两者效果非常重要。
(6)具有适当的水溶性。有效载荷必须具有适当的水溶性,以促进与抗体的偶联,并确保偶联物在生理条件下具有足够的溶解度。当过多的疏水有效载荷与抗体偶联时,产生的ADC往往会聚集并变得不稳定。此外,有效载荷的亲水性会影响亲本ADC或其代谢物的细胞膜通透性,从而影响其旁观者杀伤活性。
(7)细胞内毒性靶标。部分有效载荷为膜靶向性的,例如通过阻断离子通道或破坏凝血起作用的有效载荷,不适合用作ADC的有效载荷。目前,ADC的细胞内靶点集中在核酸或微管蛋白上。
微管是真核细胞细胞骨架的重要组成部分,在维持细胞形态、信号转导、胞内物质转运、细胞运动、细胞分裂、有丝分裂等各种细胞功能中发挥着重要作用,是肿瘤治疗的重要靶点。微管由微管蛋白组成,微管蛋白抑制剂通过与微管蛋白结合来干扰微管的动态结合,使细胞停滞在细胞分裂的G2/M期,并最终导致细胞凋亡。与大多数缓慢生长的正常细胞相比,微管抑制剂对处于快速分裂的癌细胞的毒性更强,是目前ADC药物最常用的有效载荷类型。
微管蛋白抑制剂可分为微管蛋白聚合抑制剂和微管蛋白聚合稳定剂,代表药物分别是长春碱类和紫杉烷类,已上市的ADC有效载荷多属于前者。
海兔毒素10(Dolastatin 10)是从海洋软体动物Dolabella auricularia中提取的一种线性五肽,由dolavaline(Dov)、valine(Val)、dolaisoleuine(Dil)、dolaproine(Dap)和dolaphenine(Doe)5个片段缩合而成。其作用机制通过N端与微管蛋白的长春花碱结合域,阻止其结合蛋白,抑制其介导的聚合作用;C端停留在微管蛋白可交换的三磷酸鸟苷结合位点周围,阻止其水解,从而抑制细胞的有丝分裂。小鼠模型中,海兔毒素10对L1210白血病细胞(IC50 0.01~1.00 nmol/L)、淋巴瘤细胞(IC50 0.1~1.0 pmol/L)、卵巢癌细胞(IC50 0.05~1.80 nmol/L)、结肠癌细胞(IC50 0.02~0.20 nmol/L)和肺癌细胞(IC50 0.03~0.18 nmol/L)都具有良好的抗肿瘤活性,但是对造血祖细胞也有毒性,临床上已停止单独使用该药。

图2 海兔毒素10化学结构图
其结构类似物单甲基奥瑞他汀E(MMAE)和单甲基奥瑞他汀F(MMAF)是ADC药物在研临床试验中采用最多的有效载荷。MMAE与海兔毒素10相比,C端去除了Doe的噻唑环,多了甲基和羟基,N端的叔胺变为仲胺,增加了偶联位点。MMAE结构对单抗的理化性质基本没有影响,而且保持高度稳定,在人肝脏和血浆中没有降解的迹象。MMAE作为游离的毒素,在淋巴瘤细胞中的毒性只有海兔毒素10的1/200。当其与抗体连接后,对淋巴瘤细胞的毒性能达到海免毒素10的水平。游离的MMAE表面呈电中性且通过可裂解型连接子释放,可以更容易地穿过细胞膜发挥旁观者效应。

图3 MMAE化学结构图
MMAF末端的苯丙氨酸取代带破坏了细胞膜穿透性,降低了细胞毒性和脱靶毒素的代谢速率,从而限制了显著减少了毒副作用。MMAF在淋巴瘤细胞中的毒性只有海兔毒素10的千分之一,但通过抗体进入肿瘤细胞后,活性比单独MMAF高2200倍。MMAF具有良好的亲水性,可以在一定程度上降低ADC发生聚合和沉淀的风险,而且不需要对N端进行显著修饰就可以保持原有的活性,所以临床上一般采用不可断裂的硫醚键-马来酰亚胺基已酰基(MC)将抗体与MMAF相连,提高了药物的安全性。

图4 MMAF化学结构图
Adcetris®是美国FDA批准的第一款使用奥瑞他汀类有效载荷的ADC药物,是由Seagen公司研发用于治疗CD30阳性的霍奇金淋巴瘤(HL)和系统性间变性大细胞淋巴瘤(sALCL)。该药由靶向CD30的嵌合IgG1单克隆抗体cAC10和MMAE通过蛋白酶敏感的可裂解连接子瓜氨酸-缬氨酸二肽(Val-Cit)随机偶联而成,平均药物/抗体比率(DAR)为4。

图5 Adcetris®结构示意图
美登素(Maytansine)最初是从非洲灌木Maytenus ovatus的树皮中分离出来的天然产物,是一类与微管蛋白结合并抑制微管组装的苯并大环内酯类生物碱。在体外细胞活性测定中,其IC50属于皮摩尔范围,显示出其抑制肿瘤细胞增殖的强大能力。同时还显示出良好的稳定性和溶解度。然而,由于其治疗窗口狭窄,以及由于缺乏选择性而产生的神经毒性和胃肠道反应等毒副作用,临床上已禁止直接用于治疗。然而,美登素的高细胞毒性完全满足了ADC有效载荷的要求,使美登素成为ADC有效载荷的有力候选者。
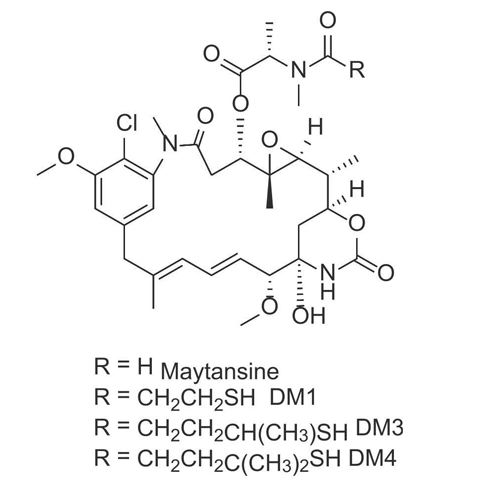
图6 美登素及其衍生物化学结构图
由于美登素没有反应性官能团,缺乏偶联位点,需要对原有结构进行修饰。对美登素类化合物构效关系(SAR)的研究已确定C3 N-酰基-N-甲基-L-丙氨酰酯侧链、C4-C5环氧化物部分、C9甲醇官能团以及C11和C13共轭双键的位置是活性的关键基团。这使得苯环和N-酰基成为化学上可修饰的实体。用3-甲基二硫代丙酰基取代美登素中的N-乙酰基,可以得到美登素衍生物DM1。DM4是通过在DM1二硫键周围添加两个甲基而获得的。DM1和DM4具有甲硫丙酰基,可以通过二硫键与连接子偶联。DM1和DM4是临床实际应用中最常用的两种美登素类有效载荷,如基于美登素衍生物获批上市的ADC药物T-SMCC-DM1。
Halichondrin B是最初从海绵Halichondraria Okadai中分离出来的聚醚大环内酯类的天然产物,被证明具有良好的抗增殖活性。其结构简化的全合成类似物甲磺酸艾日布林是一种具有抗有丝分裂作用的微管动力学抑制剂,已于2010年FDA获批用于治疗局部晚期和转移性乳腺癌(MBC)患者。

图7 甲磺酸艾日布林化学结构图
艾日布林具有独特的作用机制:它能够抑制微管聚合,不影响微管解聚,从而阻止纺锤体形成,最终导致有丝分裂停滞和随后的细胞凋亡。由于这种作用机制,艾日布林可能在紫杉烷类耐药肿瘤细胞系的情况下显示出活性。此外,艾日布林可以影响肿瘤微环境,恢复脉管系统,增加药物灌注,下调血管内皮生长因子(VEGF)和TGFβ基因的表达。这些改变可能通过减少缺氧驱动的耐药性,增加药物的肿瘤内递送,潜在地增强随后给药的治疗。
MORAb-202是由卫材研发的靶向FRα的ADC药物,由可裂解连接子Val-Cit将甲磺酸艾日布林通过半胱氨酸偶联偶联至人源化单克隆抗体farletuzumab上,DAR为4,具备旁观者效应,目前针对铂耐药卵巢癌正在Ⅱ期临床研究中。FRα(FOLR1)是一种通过GPI锚定于细胞膜的单链糖蛋白,它在正常组织中基本不表达,但在上皮来源的恶性肿瘤中过度表达,包括卵巢癌、肺癌和乳腺癌,已成为ADC药物研发的潜力靶点。
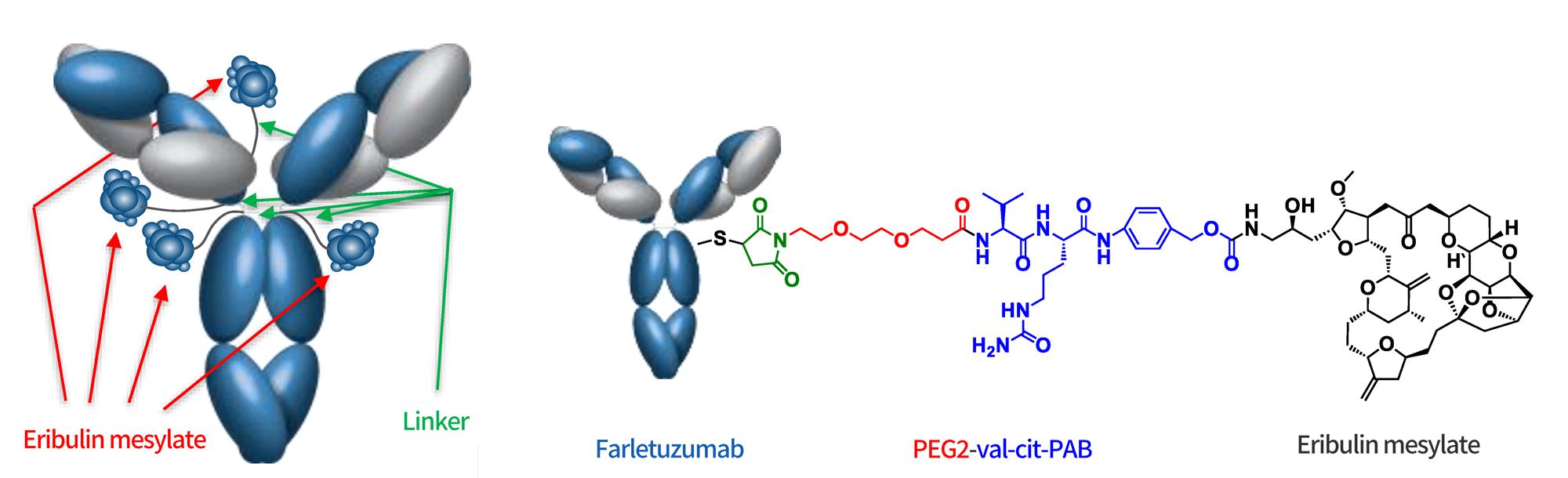
图8 MORAb-202结构示意图
Tubulysin家族是Hofle等人从黏菌培养基中分离出的天然抗有丝分裂肽,是一种线性四肽,由N-甲基-D-哌啶甲酸(D-Mep)、L-异亮氨酸(L-Ile)、Tubuvaline(L-Tuv)和Tubuphenylalanine(Tup)或Tubutyrosine(Tut)4种非天然氨基酸组成。Tubulysins可抑制微管蛋白的聚合,诱导细胞凋亡,对包括多重耐药KB-V1细胞在内的癌细胞具有较强的抗增殖活性(IC50=0.08 nmol/L),在抗癌药物开发方面具有良好的前景。

图9 Tubulysins化学结构图
对于Tubulysins的构效关系:1)Mep环是活性的关键,去掉Mep环将导致活性丧失;2)N,O-缩醛对活性影响不大,引入疏水基团可提高生物活性;改造O-乙酰基对活性影响大,优选R构型,其构型将影响微管蛋白组装;3)Tup片段的羧基甲酯化后能够提高活性,另外富电子的芳环也是活性必须的。
DXC005是由多禧生物研发的MUC1靶向ADC药物,由Tub201分子和抗MUC1单克隆抗体(IgG1)偶联而成的,Tub201分子为Tubulysin B 类似物。使用含有亲水且缓慢释放的连接子,通过连接子末端马来酰亚胺基团偶联到抗MUC1单抗体的半胱氨酸硫醇基团上。平均DAR约为4.0~4.2。拟适用的适应症为实体瘤,包括胰腺癌、结直肠癌、食管癌、卵巢癌等,目前针对胰腺癌、结直肠癌处于临床Ⅰ期。
参考文献
[1]Wang ZJ, Li HX, Gou LT, et al. Antibody-drug conjugates: Recent advances in payloads[J]. Acta Pharm Sin B, 2023, 13(10): 4025-4059. doi: 10.1016/j.apsb.2023.06.015.
[2]范啸宇,高文龙,杜子秀.auristatin类细胞毒素及其在抗体药物偶联物中的应用[J].国际生物制品学杂志, 2018, 41(5):5. DOI:10.3760/cma.j.issn.1673-4211.2018.05.010.
[3]Akaiwa M, Dugal-Tessier J, Mendelsohn BA. Antibody-Drug Conjugate Payloads; Study of Auristatin Derivatives[J]. Chem Pharm Bull (Tokyo). 2020, 68(3): 201-211. doi: 10.1248/cpb.c19-00853.
[4]周磊,张国宁,王菊仙,等.美登素类抗体药物偶联物研究进展[J].中国新药杂志, 2016, 25(22):410-419.
[5]De Sanctis R, Jacobs F, Benvenuti C, et al. From seaside to bedside: Current evidence and future perspectives in the treatment of breast cancer using marine compounds. Front Pharmacolp[J]. 2022, 8(13): 909566. doi: 10.3389/fphar.2022.909566.
[6] Furuuchi K, Rybinski K, Fulmer J, et al. Antibody-drug conjugate MORAb-202 exhibits long-lasting antitumor efficacy in TNBC PDx models. Cancer Sci. 2021 Jun;112(6):2467-2480. doi: 10.1111/cas.14898.
[7]Leverett CA, Sukuru SC, Vetelino BC, et al. Design, Synthesis, and Cytotoxic Evaluation of Novel Tubulysin Analogues as ADC Payloads. ACS Med Chem Lett. 2016, 7(11):999-1004. doi: 10.1021/acsmedchemlett.6b00274.
[8]熊樱,李英霞.海洋来源的微管蛋白聚集抑制剂tubulysins的研究进展[J].中国海洋药物, 2020, 39(2):87-92.






南京德泰生物工程有限公司 Nanjing Detai Bioengineering Co.,Ltd. ©2025 All Rights Reserved
 苏公网安备32011202001300
苏公网安备32011202001300