
在过去十年,第一代和第二代抗体药物偶联物(ADC)的经验为第三代ADC的发展铺平了道路。与第二代ADC相比,第三代ADC具有更优的连接子和偶联机制,因此具有更宽的治疗窗口。同时,第三代ADC多选择可以靶向整个细胞周期的DNA/RNA抑制剂作为有效载荷。
与微管蛋白抑制剂相比,DNA抑制剂可以通过双链断裂、烷基化/交联、嵌入等方式破坏DNA,可以作用于细胞的整个生命周期,引起细胞毒性,对实体瘤有良好的治疗效果。DNA抑制剂的靶点远少于微管蛋白抑制剂,当ADC携带相同数量的有效载荷进入细胞时,DNA抑制剂可以表现出更好的杀伤效果。以DNA抑制剂为有效载荷的ADC还可以靶向抗原表达低的肿瘤细胞,这解释了为什么DNA抑制剂在许多下一代ADC中被选为有效载荷。
拓扑异构酶Ⅰ(TopⅠ)是保证基因组稳定性和DNA结构的重要核酶,已成为ADC的热门靶标。肿瘤细胞中TopⅠ含量及活性远高于正常体细胞,抑制TopⅠ-DNA可裂解复合物解离,就能选择性抑制肿瘤细胞的快速增殖,进而杀死肿瘤细胞。TopⅠ已成为公认的抗癌药作用靶点。
TopⅠ抑制剂可分为两大类:TopⅠ毒剂和TopⅠ阻遏剂。两者都抑制活性,使DNA不能松弛,但又存在区别。TopⅠ毒剂捕获TopⅠ-DNA可裂解复合物,形成“路障”,使复制不能进行,从而导致细胞死亡,而非仅通过抑制酶催化活性杀死细胞。肿瘤细胞对TopⅠ毒剂的敏感性随TopⅠ过表达而增加,同时,TopⅠ的活性下降通常会导致TopⅠ毒剂耐受性的产生。TopⅠ阻遏剂则相反,它通过抑制酶的催化活性而杀死细胞,因此它们在TopⅠ低表达的细胞中活性更好。两类抑制剂在抗肿瘤治疗中各有优势。
目前已在临床使用或处在研发阶段的以拓扑异构酶Ⅰ为有效载荷的ADC药物主要使用的是喜树碱(CPT)及其类似物。CPT最早于1966年从我国产珙桐科植物喜树中分离得到,是一类具有五环结构的生物碱。CPT属于TopⅠ毒剂,通过与TopⅠ、DNA结合形成稳定的复合物,诱导S期细胞中的双链DNA断裂,从而导致细胞凋亡(图1)。然而,CPT极低的溶解度阻止了其作为癌症治疗药物的广泛使用,故改造重点放在改善喜树碱理化性质、提高其内酯环稳定性等方面。近年来已合成了一系列喜树碱类似物,如已上市的10-羟基喜树碱(HCPT)、拓扑替康(Topotecan,TPT,图2)、伊立替康(Irinotecan,CPT-11)和贝洛替康(Belotecan,BLT)。
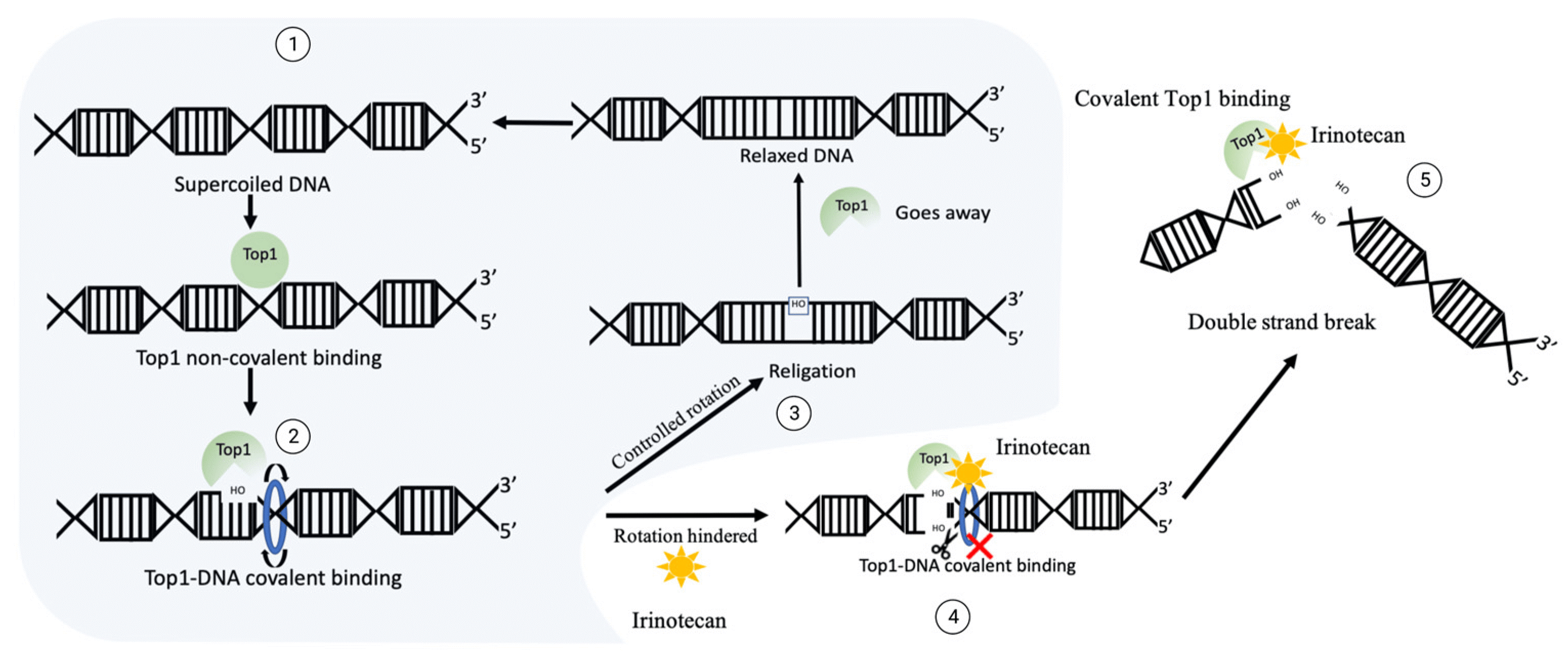
图1 喜树碱类毒素CPT-11暴露情况下DNA断裂机制
正常情况下TopⅠ的作用机制①TopⅠ感应超螺旋并形成切口使得②DNA松弛③TopⅠ重新连接切口并离开位点。
在CPT-11暴露的情况下,TopⅠ与DNA共价交联④3′端游离-OH的连接被阻断,并转化为⑤双链断裂并促进细胞毒性。
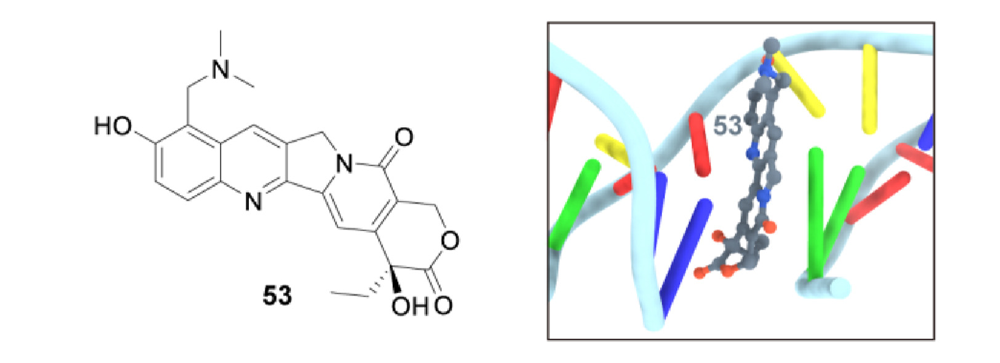
图2 拓扑替康结构及与TopⅠ-DNA复合物结合模式图
拓扑替康与DNA间为堆积作用,与TopⅠ的Asp-533以氢键结合,与磷酸酪氨酸和Asn-722之间的活性位点以水桥形式相互作用。
SN-38是CPT-11的活性代谢产物,CPT-11 C10位双哌啶基侧链的氨基甲酸酯键可被肝脏中的羧酸酯酶裂解,从而产生SN-38(图3)。对CPT-11和SN-38构效关系进行研究发现:(1)C10位以-OH取代的喜树碱是高效低毒化合物;(2)A环用较大基团取代后活性降低;(3)A和B环的刚性平面结构对喜树碱生物活性有重要影响,可使活性增强;(4)C5位以-OH,-OCOCH3和-C2H5等取代后活性完全消失,可能是位阻增加,阻碍B环DNA-酶复合物嵌合;(5)C7和C9位为极性取代基时,可使活性增强;C7位以乙基取代活性最强;(6)C20位羟基必须为α位,为药物活性必需基团;(7)完整的内酯环(E环)或酯基是维持药物活性的重要基团;(8)在C9、C10、C11位加上亲水性羟基、硝基基团可以增加药物的水溶性。
SN-38对几种不同来源的人类癌细胞系的IC50约在1.0~6.0 nM范围内。在体内生理pH条件下,SN-38会因pH微弱升高打开作为主要活性基团的内酯环,从活性较强的内酯环形式水解成活性较低的羧酸盐形式,导致效力降低;C10位会快速通过UGT-1A(尿苷二磷酸葡萄糖醛酸基转移酶)被葡萄糖醛酸化。
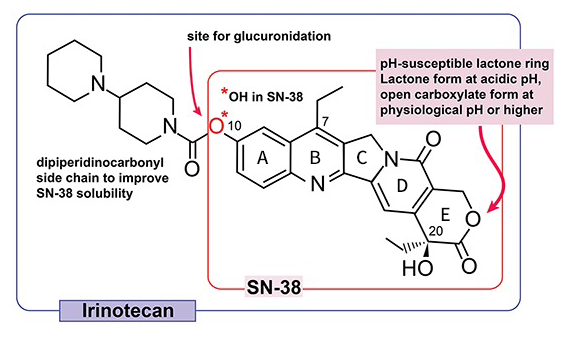
图3 CPT-11、SN-38化学结构图
Trodelvy®是由Immunomedics研发一款靶向Trop-2 ADC药物,于2020年4月FDA获批上市。Trodelvy®通过CL2A连接子将靶向癌细胞表面Trop-2的人源化单克隆抗体hRS7和与SN-38偶联,平均DAR为7.6,具有旁观者杀伤效应。CL2A连接子具有短的PEG(聚乙二醇)残基增加溶解度,减少聚合的可能;加入马来酰亚胺基团,使连接子-有效载荷复合物与抗体上产生的巯基反应,形成稳定的硫醚键。CL2A和SN-38通过pH敏感的碳酸苄酯键结合,在低pH环境中(肿瘤的微环境)SN-38可以从连接子中释放。目前,Trodelvy®全球获批适应症包括局部晚期或转移性乳腺癌及局部晚期或转移性尿路上皮癌。

图4 Trodelvy®结构示意图
依喜替康甲磺酸盐(Exatecan mesilate, DX-8951f)是一种水溶性CPT衍生物,与其他CPT类似物相比,具有更强Top-Ⅰ抑制和抗肿瘤活性。DXd为依喜替康衍生物,具有高膜渗透性,IC50 2.05~17.8 nM,可克服P-糖蛋白介导的多药耐药性,对各种肿瘤移植模型(包括体内CPT-11耐药肿瘤)具有更强的疗效。
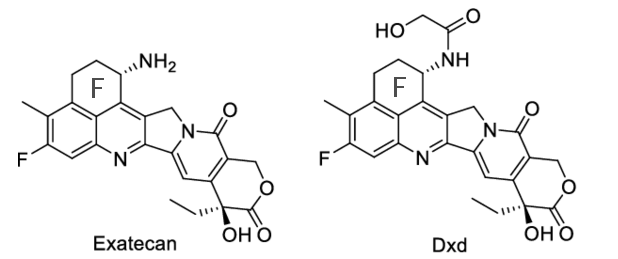
图5 Exatecan、DXd化学结构图
Exatecan有一个F环,其中包含第二个手性中心,使其合成和衍生化复杂化。构效关系研究表明,C11位的氟取代通常会使细胞毒性增加数倍,并且C20中心必须处于S构型才能使分子保持活性。其内酯环稳定性的提高归因于C11氟取代基和F环的作用。
在链霉菌中发现的吡咯并[2,1-c][1,4]苯二氮卓类(PBD)是一类具有抗肿瘤活性的天然产物,通过鸟嘌呤与PBD的亲电性N10/C11亚胺形成共价键,将DNA小沟选择性烷基化,导致DNA链间交联,使细胞周期停滞在G2/M期,引起细胞凋亡。
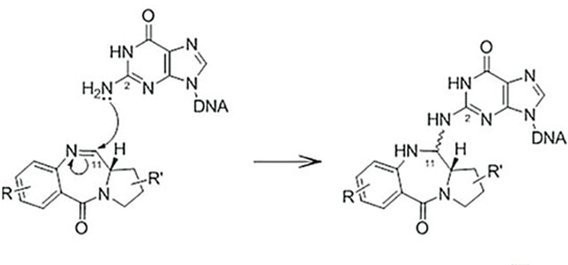
图6 PBD与DNA共价结合的机制
PBD二聚体具有用作有效载荷的潜力,其与DNA形成的链间交联的扭曲最小,可以有效逃避DNA的修复机制,使其在细胞中保持持久且较高的生物活性。PBD二聚体对各种癌细胞类型表现出皮摩尔范围内的细胞毒性,属于高毒素有效载荷,一般选择搭配较低DAR值(通常在2左右)。这种强效力提供了靶向低拷贝数抗原的能力,这对实体瘤的治疗特别重要。其次,PBD二聚体具有较好的膜通透性,已被证明在体外/体内均可以产生有效的旁观者杀伤效应。PBD二聚体的半衰期非常短,这确保了游离药物的脱靶毒性在全身的蓄积受到限制。许多有效的PBD二聚体载荷不是显著的P-糖蛋白底物,这使它们较长春碱类、紫杉烷类、蒽环类、拓扑异构酶抑制剂等P-糖蛋白底物具有重要的优势。除以上优势以外,PBD二聚体还拥有较为简单的合成途径。
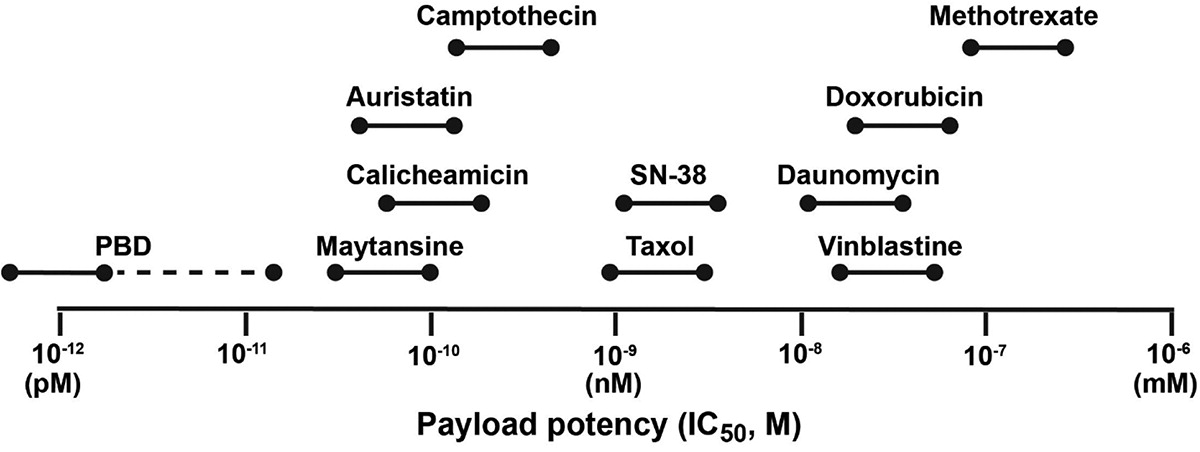
图7 各类有效载荷IC50一览
ZynlontaTM由ADC Therapeutics研发的靶向CD19 ADC药物,于2021年4月获FDA加速批准上市,用以治疗复发性或难治性弥漫性大B细胞淋巴瘤(r/r DLBCL)。由靶向CD19的人源化IgG1ҡ单克隆抗体通过马来酰亚胺连接子偶联到Tesirine(SG3249),Tesirine由8个聚乙二醇(PEG8)、组织蛋白酶B可裂解的Val-Ala连接子以及PABA间隔子组成,可释放PBD二聚体有效载荷SG3199。
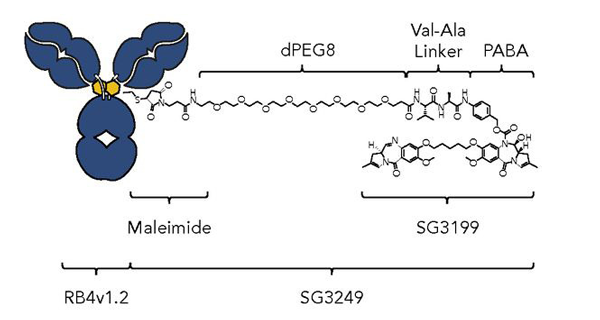
图8 ZynlontaTM结构示意图
CS5001是以PBD为前药(pPBD)设计的靶向ROR1 ADC药物,在多种表达ROR1的肿瘤细胞系中显示出很强的选择性,并在血液和实体瘤异种移植小鼠模型中显示出显著的体内抗肿瘤活性。临床试验目前正在进行中。
ADC领域的一个明显趋势在于适应症正从血液瘤转向实体瘤,这种趋势可能使对有效载荷的选择从靶向微管抑制剂转向靶向DNA损伤剂。后者通常表现出更高的效力,能满足新一代ADC药物所需的治疗指数。此外,使用DNA损伤剂作为有效载荷,可以选择表达水平相对较低的肿瘤特异性/相关表面受体。使用DNA损伤剂作为ADC有效载荷的技术已经相对成熟,然而DNA损伤难以修复,对其带来的毒副作用也应相对慎重。目前,只有一种以DNA损伤剂为有效载荷的ADC药物在临床上使用。未来,针对安全性/耐药性问题仍需进一步研究。
以上载荷对部分肿瘤仍无法产生抑制作用,或易产生耐药性。生长缓慢的肿瘤细胞通常不像快速分裂的那样依赖微管蛋白介导的细胞过程。因此,为了进一步扩大ADC有效载荷的范围,确定对快速/慢速增殖细胞均有效并能逃避多药耐药性的有效载荷,人们的注意力转向了靶向RNA的有效载荷。靶向RNA的小分子抑制剂可以同时作用于分裂期和休眠期的肿瘤细胞,有望解决由于肿瘤细胞休眠引起的肿瘤耐药性和复发扩散的问题。目前有两种主要类型的RNA抑制剂可用于ADC有效载荷:RNA剪接抑制剂(泰兰斯他汀及其类似物)和RNA聚合酶Ⅱ抑制剂(鹅膏毒肽)。
RNA剪接通过剪接内含子和外显子将RNA转化为mRNA来控制代谢、血管生成、癌细胞增殖和转移。它可以直接控制转录的起始、延伸和终止,可以作为癌症抑制的生物学靶点。研究发现,在RNA剪接调节药物的干扰下,肿瘤细胞可以产生新抗原,通过MHCⅠ呈递为新抗原表位,从而刺激抗肿瘤免疫。RNA剪接调控可作为肿瘤抗原的潜在来源,具有应用于肿瘤免疫检查点治疗的潜力。
泰兰斯他汀是一种从布鲁氏菌中分离出来的天然产物,通过与剪接体U2-snRNA亚复合物SF3b结合而激活。泰兰斯他汀家族通过抑制真核mRNA剪接途径,对剪接体具有很强的结合和抑制作用,对多种癌细胞系的IC50处于低纳摩尔,有潜力用作ADC药物的有效载荷。
辉瑞曾探索使用泰兰斯他汀及其类似物作为抗体药物偶联物有效载荷,将含羧酸的泰兰斯他汀半合成类似物直接偶联至曲妥珠单抗表面赖氨酸。该ADC对HER2高表达胃癌细胞系N87和MDR1过表达细胞系有效,且在低至1.5mg/kg的剂量下,对胃癌异种移植模型也能产生优于临床批准的T-DM1的效力。
鹅膏毒肽是一类双环八肽毒素,属于真核RNA聚合酶Ⅱ(RNAPⅡ)的选择性抑制剂,可导致细胞凋亡。鹅膏毒肽中研究最为深入的是α-鹅膏蕈碱(α-amanitin),这种毒素具有突破耐药性、破坏沉默期肿瘤细胞的潜力,可以有效防止肿瘤转移和复发。对α-鹅膏蕈碱晶体结构的研究发现,抑制剂的位置与桥螺旋(bridge helix)相邻,启动环主要处于远离加成位点的稳定构象中(图9)。α-鹅膏蕈碱环肽骨架形成一个口袋,允许与RNAPⅡ亚单位Rpb1 His1085侧链咪唑环形成特异性互补氢键,从而对启动环产生了一个约束位点。α-鹅膏蕈碱对咪唑的特异性主要通过Gly7的酰胺NH与Rpb1 His1085 N(D1)和Asn1的羰基与Rpb1 His1085 NH(E2)的相互作用来实现。
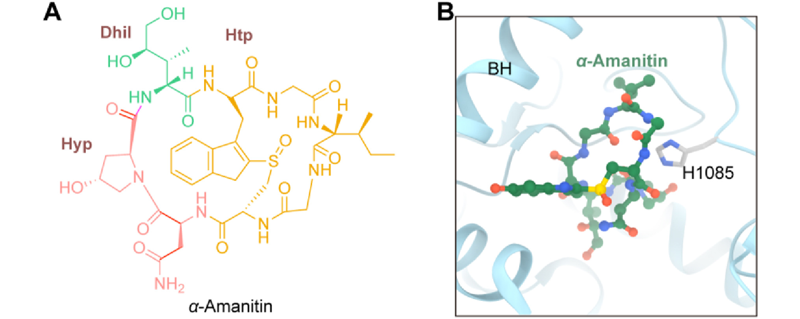
图9 α-鹅膏蕈碱结构及与RNA结合模式图
由于鹅膏毒肽分子量小、水溶性好、非P-糖蛋白底物以及对RNAPⅡ的抑制作用,近年来作为有效载荷在癌症研究中受到越来越多的关注。HDP-101是德国Heidelberg Pharma开发的以鹅膏毒肽为有效载荷的,靶向B细胞成熟抗原(BCMA)ADC药物,目前正处于Ⅰ/Ⅱa期临床试验中,用于治疗复发/难治性多发性骨髓瘤。
以泰兰斯他汀和鹅膏毒肽为有效载荷的ADC药物能够避免治疗耐药性,对休眠的肿瘤细胞也产生作用,现有的标准疗法很少能达到这种效果。虽然暂时没有使用靶向RNA有效载荷的ADC药物上市,但它们的巨大潜力值得进一步探究。
参考文献
[1]Wang ZJ, Li HX, Gou LT, et al. Antibody-drug conjugates: Recent advances in payloads[J]. Acta Pharm Sin B, 2023, 13(10): 4025-4059. doi: 10.1016/j.apsb.2023.06.015.
[2]Kumar S, Sherman M Y. Resistance to TOP-1 Inhibitors: Good Old Drugs Still Can Surprise Us[J]. Int. J. Mol. Sci, 2023, 24: 7233.
[3]Goldenberg DM, Cardillo TM, Govindan SV, et al. Trop-2 is a novel target for solid cancer therapy with sacituzumab govitecan (IMMU-132), an antibody-drug conjugate (ADC)[J]. Oncotarget, 2015, 6(26): 22496-22512.
[4]王丽焱.抗肿瘤药伊立替康的研究进展[J].国外医学药学分册, 2004.
[5]Goldenberg DM, Sharkey RM. Antibody-drug conjugates targeting TROP-2 and incorporating SN-38: A case study of anti-TROP-2 sacituzumab govitecan[J]. mAbs, 2019, 11(6): 987–995.
[6]Mantaj J, Jackson PJ, Rahman KM, et al. From Anthramycin to Pyrrolobenzodiazepine (PBD)-Containing Antibody-Drug Conjugates (ADCs). Angew Chem Int Ed Engl. 2017, 56(2):462-488.
[7]Hartley JA. Antibody-drug conjugates (ADCs) delivering pyrrolobenzodiazepine (PBD) dimers for cancer therapy[J]. Expert Opin Biol Ther, 2021, 21(7):931-943.
[8]Zammarchi F, Corbett S, Adams L, et al. ADCT-402, a PBD dimer-containing antibody drug conjugate targeting CD19-expressing malignancies[J]. Blood, 2018, 131(10):1094-1105.
[9]https://mp.weixin.qq.com/s/Os3fgrcjy0W1dGP2uTLASw






南京德泰生物工程有限公司 Nanjing Detai Bioengineering Co.,Ltd. ©2025 All Rights Reserved
 苏公网安备32011202001300
苏公网安备32011202001300