
PCR技术具有重复性好、灵敏度高、快速简便等突出优点,是科研研究最常用到的技术之一。但是由于灵敏度高,实验操作人员细微的操作失误就可能导致产生污染或没有扩增出目的条带,因此PCR实验对操作有一定的要求。本文对PCR实验操作中的注意事项、PCR实验污染问题及预防措施做一介绍。
PCR实验中一个令人头痛的问题是易污染,极其微量的污染就可造成假阳性结果。导致污染的原因有:
样本污染:收集标本的容器被污染;标本放置时,由于密封不严溢于容器外;容器外沾有标本造成交叉污染
试剂污染:在PCR试剂配置过程中,由于加样器、容器、双蒸水及其它溶液被PCR核酸模板污染;
PCR产物污染:最主要的污染源。如果在PCR实验过程中形成了气溶胶(在空气与液体面摩擦时、在操作时比较剧烈的摇动反应管,开盖时、吸样时及污染加样器的反复吹打都可形成气溶胶,一个气溶胶可含48000拷贝),则气溶胶可能对之后进行PCR扩增实验产生污染。
PCR技术实际上就是在模板DNA、引物、dNTP都存在的条件下依赖于DNA聚合酶的酶促反应,其效率和特异性取决于两个方面:一是引物与模板的特异结合;二是多聚酶对引物的有效延伸。因此,引物的设计对于PCR极为重要,引物设计的不好,会导致无法顺利扩增得到目的片段。
本文主要对PCR引物设计的原则,常见的引物设计工具及引物设计的总体流程做一介绍。
设计PCR引物的目的是为了找到一对合适的核苷酸片段,使其能有效地扩增模板DNA序列。根据多年来的实践经验,引物的设计有一些大家都认可的原则。
引物设计有3条基本原则:首先引物与模板的序列要紧密互补,其次引物与引物之间避免形成稳定的二聚体或发夹结构,再次引物不能在模板的非目的位点引发DNA聚合反应(即错配)。具体实现这3条基本原则需要考虑诸多因素,如引物长度,GC含量,引物碱基分部,Tm值,引物特异性,形成引物二聚体及发夹结构的能值等等。
目前引物的设计主要借助一些分子生物学软件或在线工具来实现。常见的引物设计软件有Oligo 6、Premier Premier、Primer 3、Vector NTI Suit、Dnasis、Omiga、Dnastar等。最为常用的是Oligo 6、Premier Premier及Primer 3。
Oligo是一款非常著名的软件,可以实现引物的设计及引物的评估分析,是目前最好、最专业的引物设计软件。其主要功能包括:普通引物对的搜索、测序引物的设计、杂交探针的设计以及评估引物对质量等等。
PremierPremier是由加拿大的Premier公司开发的专业用于PCR或测序引物以及杂交探针的设计和评估的软件,也是一款应用很广泛的软件。利用该软件还可以实现简并引物的设计。
Primer 3是由美国whitehead生物医学研究所基因组研究中心在因特网上提供的一款免费的在线PCR引物设计程序(在线网址:http://primer3.ut.ee/),一个非常简单却高效的引物设计在线软件。只需在目标序列中粘贴DNA序列后点击搜索即可。其中,可通过多种方式来对结果进行筛选,包括PCR产物的大小、引物大小、Tm范围和其它参数。同样,还可使用Primer3来设计用于PCR-ELISA的杂交探针和其他基于探针的PCR引物设计。
运用软件Premier Premier或Primer 3来设计引物,再用软件Oligo6进行引物评估,就可以初步获得一组比较满意的引物。
引物的二次筛选是指在初次筛选出的几对引物中进一步筛选出合适我们进行特异、高效PCR扩增的那对引物。本步骤应注意一下两点,一是得到一系列引物分别在Genebank中进行回检。也就是把每条引物在对比工具(http://www.ncbi.nlm.nih.gov/blast/)的blastnr中进行同源性检索,弃掉与基因组其它部分同源性比较高的引物,也就是有可能形成错配的引物。一般连续10bp以上的同源有可能形成比较稳定的错配,特别是引物的3’端应避免连续5-6bp的同源。二是以mRNA为模板设计引物时要先利用生物信息学的知识大致判断外显子与内含子的剪切位点,然后弃掉正好位于剪切位点的引物。
引物确定以后,可以对引物进行必要的修饰,例如可以在引物的5′端加酶切位点序列;标记生物素、荧光素、地高辛等。
经过初次筛选和二次筛选后得到的那对引物便可以用于合成,合成后我们经过PCR扩增可以对引物进行最终的评估。一是PCR扩增的特异性和效率。经过PCR条件优化后能否获得特异性条带,即无目的条带之外的多余条带。另外,PCR产物的量是否足够,即无不出带和条带很弱的现象。二是以DNA为模板设计引物时,PCR扩增产物是否与预期PCR产物大小相当。如果差距太大(大于100bp),有可能是错配产物,三是是否形成引物二聚体带。
我们结合引物最终评估和测序的结果可以对引物设计的成败做出判定,为我们以后进行引物设计积累宝贵的经验。
在PCR扩增完成后,反应体系中除了DNA片段,还存在离子、dNTP、引物及聚合酶等物质,这些物质会对后续的实验(克隆、测序等)产生不利的影响,需要对产物进行纯化回收。回收DNA片段有两种途径,即直接回收和从凝胶中回收,每种纯化途径都有相对应的试剂盒。
在PCR扩增完成后进行琼脂糖凝胶电泳检测,在条带单一无其他杂带的情况下,可以用产物纯化试剂盒对PCR扩增产物直接进行纯化。目前市面上的产物纯化试剂盒大多是利用吸附柱的方法,其实验过程为“吸附-洗杂-洗脱”:将PCR产物置于DNA纯化柱中,产物中DNA片段会吸附于DNA纯化柱上,利用wash buffer通过一系列快速漂洗-离心的步骤,将引物、核苷酸、蛋白、酶等杂质去除(洗杂需重复多次以尽可能的洗去杂质,提高产物纯度),最后用洗脱液将DNA片段洗脱。
如果电泳检测结果存在非特异性条带,则需要通过切胶将目的条带分离出来,随后利用胶回收试剂盒对凝胶回收纯化。凝胶回收纯化与产物直接纯化相比多了一个溶胶的过程,两者纯化原理基本相同。
产物直接纯化的回收率比胶回收高,但只适用于电泳结果为单一条带的情况。凝胶回收纯化需要先跑胶然后将目的条带切胶回收,纯化产物更纯净。
PCR产物纯化回收有两个重要的技术指标:纯度和回收率。回收率不理想会使工作量大大的增加,下面就介绍一些能够提高产物回收率的小技巧:
常规的PCR技术只能对两端序列已知的DNA片段进行扩增,但在实际工作中常常需要通过基因上一小段已知序列对其邻近序列进行扩增。为了解决这一问题,科学家们对常规PCR进行技术改进,发明了多种获得已知基因两侧序列的方法,包括反向PCR、锚定PCR、RACE-cDNA末端的快速扩增等。本文主要对反向PCR做一介绍。反向PCR(inverse-PCR,IPCR)具有与常规PCR方向相反的引物,并且IPCR的扩增方向与普通PCR相反,因此称为反向PCR。
IPCR实验是将待扩增片段环化,通过一对方向相反的引物实现已知序列两侧基因序列的扩增(见下图)。常规PCR的方向是相对的,可以扩增一对引物之间的片段,因此想要扩增引物两侧的基因片段,需要设计方向相反的引物。但用方向相反的引物进行PCR扩增,是无法得到足够的产物的,因为每个引物只能对各自的模板线性扩增,无法进行指数级增长。为了解决这个问题,需对扩增的DNA模板先进行酶切,然后连接环化,使其引物方向成为相对,而后再进行PCR扩增。

图1:反向PCR技术原理
引物的方向与常规PCR相反,其设计原则与常规PCR相同。
取基因组DNA,先用适宜的限制性酶进行切割,随后去除反应中的限制性酶,再用连接酶将酶切片段进行自身连接,形成一环状结构(为了提高IPCR效率,有时会将环化模板再次线性化会有利于扩增,即在已知序列的引物之间寻找一合适的酶切位点,且该位点在待研究的侧翼未知序列上不存在,用相应的限制性酶消化,即可获得线性化的模板)。
根据模板设置合适的条件进行PCR扩增。一般来说,在IPCR实验中只进行一轮PCR是不够的,通常会设计不同的引物进行两轮或两轮以上的PCR反应(巢式PCR)。
扩增产物可以通过琼脂糖凝胶电泳分析判断其分子量,也可以进行TA克隆并进行测序获得基因序列。
反转录PCR对克隆侧翼未知DNA序列快速有效,是常规PCR技术的重要补充,是分子生物学技术的重要方法之一。IPCR在分子生物学研究中应用广泛,可以检测病毒、转座子[1]等在基因组中的整合位点,克隆基因的邻接序列以及建立基因组步移[2]文库等。虽然该技术目前存在一定的缺陷(如基因组的复杂度、扩增的长度等有一定的限制),但随着分子生物学技术的发展,必将会得到不断的完善。
注释:
[1] 转座子:基因组中一段特异的具有转位特异性的独立DNA序列,可以从基因组中单独复制或断裂下来,环化后插入另一位点,并对其后的基因起调控作用。
[2] 基因组步移:通过一段已知序列探知其旁邻的序列,并将探得的序列作为已知序列进一步探知其旁邻序列的方法称为基因组步移。
尽管目前PCR技术已相当普遍和成熟,但由于PCR反应中许多条件(如Mg2+、dNTPs、引物、模板、循环中的参数等)仍然会影响实验结果,特别对于一些复杂的基因组DNA模板,普通PCR往往存在非特异性扩增,得不到理想的产物。为了解决PCR非特异性扩增的问题,Don等人于1991年发明了降落PCR(touchdown PCR,TD-PCR)技术。
降落PCR是通过对反应体系中退火温度进行优化来提高反应的特异性,其基本原理为:根据引物的Tm值,设置一系列从高到低的退火温度,开始选择的退火温度高于估计的Tm值,每一个(或n个)循环降低1℃(或n℃)退火温度,随着循环的进行,退火温度逐渐降低至Tm值,最终低于Tm值达到一个较低的退火温度。最后以此退火温度进行10个左右的循环。
PCR反应中退火温度会对扩增结果产生影响,随着退火温度的升高,扩增特异性会变好,同时扩增效率会变低。退火温度过高会使PCR效率过低,退火温度过低则会使非特异性扩增过多。降落PCR一开始先用高温扩增,保证扩增的严谨性(在较高的退火温度下通常得到特异性扩增产物),待目的基因的丰度上升后,降低扩增的温度,提高扩增的效率。当退火温度降到非特异扩增发生的水平时,特异产物会有一个几何级数的起始优势,在剩余反应中非特异的位点由于丰度低无法和特异位点竞争,从而产生单一的占主导地位的扩增产物。
降落PCR具有以下三个特点:
通常降落PCR的退火温度范围可跨越15℃,从高于Tm值几度到低于其10℃左右,在每个温度上循环1~2个周期,然后在较低的退火温度上循环10个周期左右。
人们为了简化实验流程,同时避免最低退火温度过低会有非特异性产物出现的可能性,将TD-PCR退火温度缩短至10个系列温度,此扩增方法称作MTD-PCR。后来人们又将MTD-PCR再次改进,将退火温度缩短至5甚至4个温度范围,并将这种方法称为SMTD-PCR。改良后的TD-PCR在每一个停留的退火温度上循环4或者5次,最后一个退火温度循环次数增加至20~30个循环,以最大可能提高其特异产物与非特异产物的比率。但改良后的TD-PCR需要从较低的退火温度开始降落,因为尽管Taq等酶耐热,但经过太长时间的高温后其活性也会下降,会由于DNA聚合酶活性丧失过多而得不到足量的PCR产物。
如果扩增后跑胶观察到许多产物带或高分子量成片产物,可采取以下措施:
降落PCR与温度梯度PCR都是对反应体系中的退火温度进行优化,但两者在原理上有所不同。
温度梯度PCR是指在退火温度不太明确时,为了找到最优退火温度,在一台PCR仪上同时做多管PCR,每一管放在仪器内不同列(有的是不同行)上,分别进行PCR(如50-60℃可以分6管,分别50℃,52℃,54℃,56℃,58℃,60℃),最终找到最合适的退火温度,并进一步以此退火温度进行普通PCR扩增。
降落PCR是为了提高反应的特异性,通过设计一系列不断降低的退火温度,在同一PCR管内进行PCR扩增,最终获得大量的特异性扩增产物。
与温度梯度PCR相比,降落PCR更有优势,因为采用梯度PCR选择合适的退火温度时需要多次反应或多管反应,并且即使通过多次试验找到最佳的退火温度后,在更换其它的PCR仪进行同样的扩增时最适的退火温度也有可能发生改变,需要重新进行最佳温度的摸索。而降落PCR只需一次反应就可以获得很好的扩增效果,避免了对每对引物进行最佳复性温度的优化和测定工作,并且降落PCR在很大程度上削弱了仪器性能对扩增效果的制约。
降落PCR适用于经常更换引物的实验,在不知道Tm值,同时不想很麻烦的找出最佳Tm值的情况下,使用降落PCR可以快速、特异的得到目的扩增片段。现在很多PCR仪具有设置降落PCR的程序,降落PCR在研究领域中已得到了广泛的应用。根据各种不同工作的需要,降落PCR还可与其它PCR方法同时使用,如实时定量降落PCR、竞争降落PCR等,这些联合PCR的反应体系,所需材料均与实时定量PCR和竞争定量PCR相同,所不同的是在PCR的循环过程中使用退火温度渐低的降落PCR方法,检测PCR产物的方法与硬件也均同于实时定量PCR和竞争定量PCR。
实时荧光定量PCR(Realtime fluorescence quantitative PCR,qPCR),它是指在PCR反应体系中加入荧光基团,利用荧光信号累积实时监测整个PCR进程,最后通过标准曲线对初始模板进行定量分析的方法。该技术于1996年由美国Applied Biosystems公司推出,它的出现解决了传统PCR不能对初始模板定量分析的问题。荧光定量PCR具有准确性高、灵敏度高、特异性强等优点,目前已广泛应用于分子生物学研究和医学研究等领域。
在PCR反应体系中加入荧光基团,随着PCR反应的进行,PCR反应产物不断累积,荧光信号强度也等比例增加。每经过一个循环收集一个荧光强度信号,通过荧光强度变化检测产物量的变化,最终得到得到一条荧光扩增曲线图,扩增曲线横坐标表示循环数,纵坐标表示荧光强度。
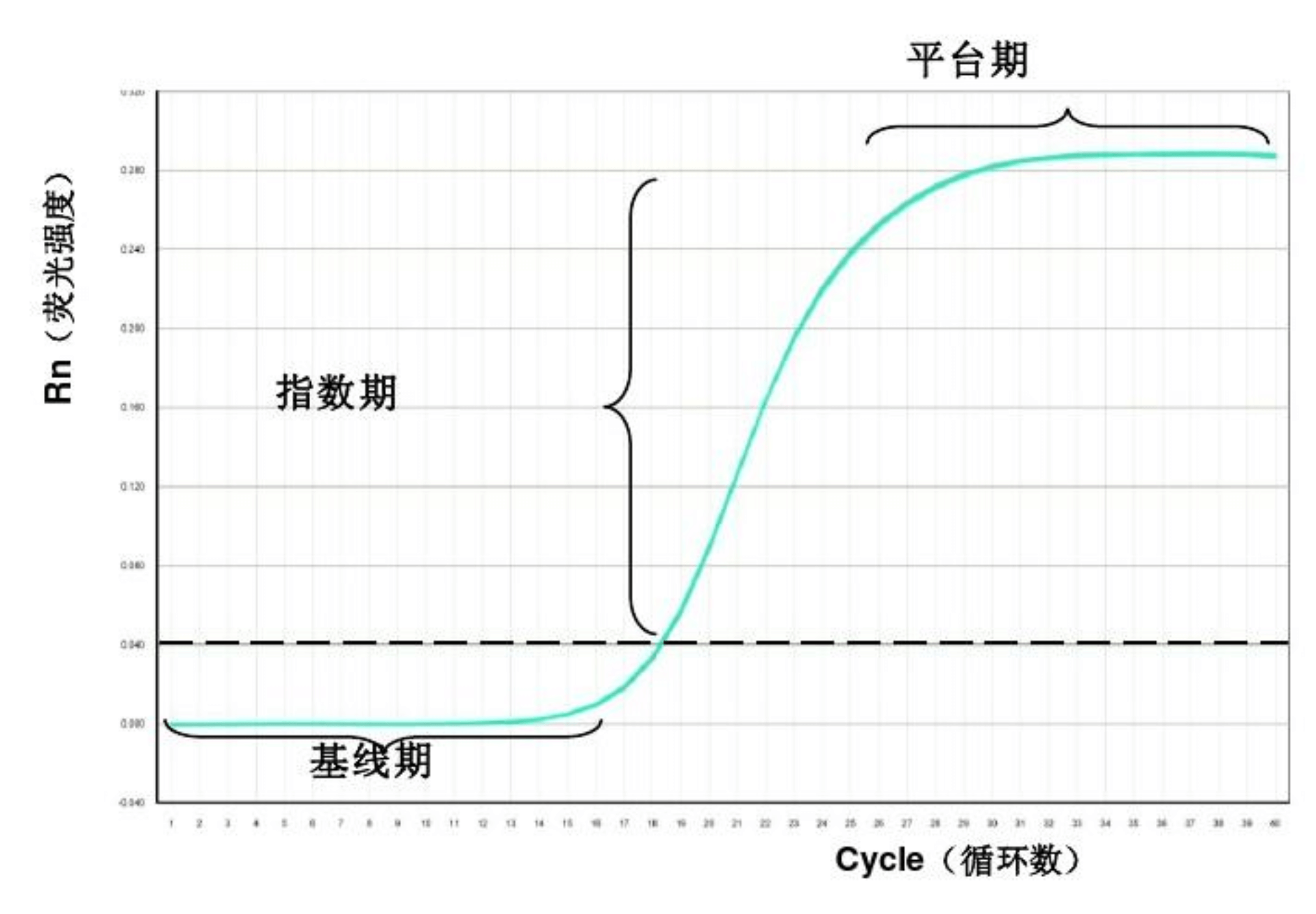
图1:qPCR扩增曲线
一般而言,荧光扩增曲线可以分为三个阶段:荧光背景信号阶段(基线期)、荧光信号指数扩增阶段和平台期。在荧光背景信号阶段,扩增的荧光信号被荧光背景信号所掩盖,无法判断产物量的变化;在平台期,扩增产物已不再呈指数级的增加,PCR的终产物量与起始模板量之间没有线性关系,根据最终的PCR产物量也不能计算出起始DNA拷贝数;只有在荧光信号指数扩增阶段,PCR产物量的对数值与起始模板量之间存在线性关系。
在了解荧光定量PCR如何对初始模板进行定量之前,需要了解几个在荧光定量PCR分析中常用的术语:
利用实时荧光定量PCR对样品的初始模板量进行定量分析,需要利用已知起始拷贝数的标准品作出标准曲线,再通过荧光定量PCR获得未知样品的Ct值,最后从标准曲线上计算出该样品的起始拷贝数。
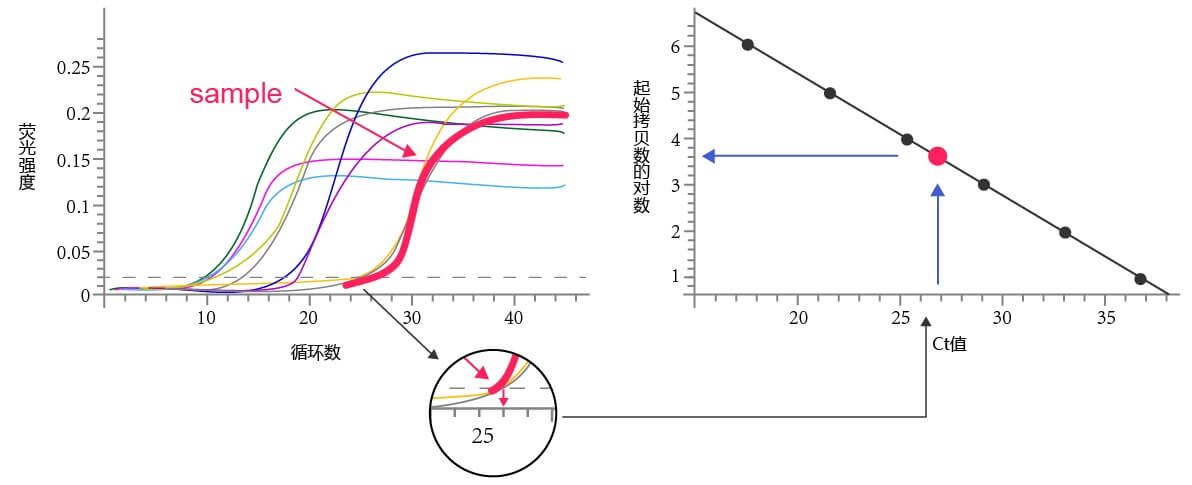
图2:对初始模板进行定量分析
设计特定引物,利用PCR对目的基因片段进行扩增,随后将目的基因片段克隆至载体中,测序验证是否为阳性重组质粒,最后收集阳性克隆质粒作为标准品。
测定质粒的浓度,依据公式计算出质粒的拷贝数。对质粒进行系列稀释,分别作为模板进行荧光定量PCR并记录Ct值。最后依据起始拷贝数的对数及Ct值绘制标准曲线,得到标准方程。当需要对起始模板进行定量时,只需要得到扩增曲线,读得Ct值,带入标准方程即可对起始模板定量。
实时荧光定量PCR荧光标记方法可分为荧光染料法和荧光探针法两类。染料法是利用荧光染料可以嵌合到DNA双链内部的特性,来指示扩增产物的增加。而探针法是利用与靶序列特异结合的荧光探针的信号积累来指示扩增产物的增加。
染料法是利用荧光染料可以嵌合到DNA双链内部的特性,来指示扩增产物的增加。现以最常用的SYBR GreenⅠ为例,介绍染料法荧光定量PCR的工作原理:
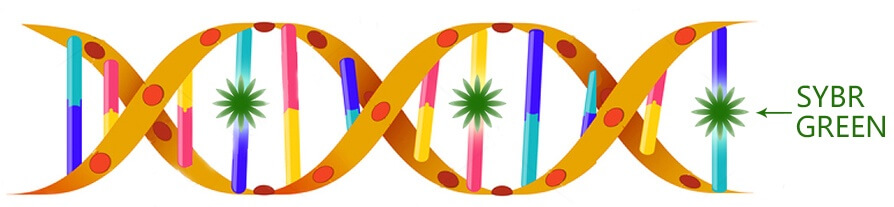
图3:荧光染料法原理
SYBR GreenⅠ是一种荧光染料,可嵌合到双链DNA的内部。在游离状态下,SYBR Green Ⅰ发出微弱的荧光,但一旦与双链DNA结合,其荧光增加1000倍。一个反应发出的全部荧光信号与出现的双链DNA量呈正比,随扩增产物的增加而增加,可通过荧光信号的积累来指示扩增产物的增加。
探针为一段寡核苷酸,可与DNA序列特异性结合,一个模板结合一个探针。探针5’端具有报告基团(R),可发光;3’端有荧光淬灭基团(Q),能吸收光。探针完整时R基团所发射的荧光能量被Q基团吸收,PCR仪检测不到荧光信号。随着扩增的进行,探针会被聚合酶水解,报告基团发出的光没有被淬灭基团吸收,从而被仪器检测到。每扩增一条DNA链,释放一个荧光信号,实现了荧光信号的累积与PCR产物形成完全同步。
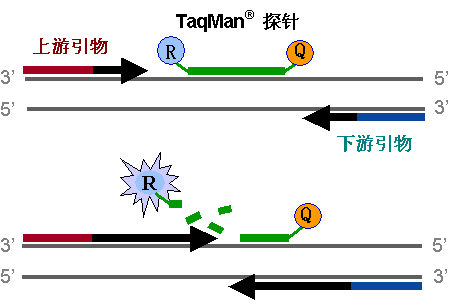
图4:探针原理
目前已经开发出来的探针有Taq Man探针、Taq man MGB探针、双杂交探针、分子信标、Lux杂交探针、Simple proble探针等。
探针法与染料法对比
| 探针法 | 染料法 | |
|---|---|---|
| 优点 |
|
|
| 缺点 |
|
|
实时荧光PCR技术已广泛应用于临床及生命科学研究的各个领域中。在科研方面可定量分析各种基因的表达分析,基因突变和多态性分析,单核苷酸多态SNP测定及易位基因的检测;在医疗方面可用于免疫组化分析、临床疾病早期诊断、病原体检测、耐药性分析、肿瘤微小残留病变研究等。
[1]Mikael KA,Jose MA,Martin B, et al. The real-time poly-merase chain reaction. Molecular Aspects of Medicine[J],2006,27:95-125;
[2]纪冬,辛绍杰等. 实时荧光定量PCR的发展和数据分析[J].生物技术通讯,2009,20(4):598-600;
[3]陈旭,齐凤坤等. 实时荧光定量PCR技术研究进展及其应用[J].东北农业大学学报,2010,41(8):148-155;
聚合酶链式反应(Polymerase chain reaction,PCR)是一种分子生物学技术,用于体外扩增特定的DNA片段。PCR技术主要过程是,通过人工合成的一小段单链DNA片段(引物)与模板DNA特定的区域特异性结合,以四种dNTP为底物,通过DNA聚合酶沿着引物和模板形成的双链部分的3’端聚合形成DNA片段,实现DNA体外扩增。

PCR技术的发明使微量的核酸操作变得简单易行,同时使核酸研究脱离于活体生物,极大地推动了分子生物学以及生物技术产业的发展。随着对PCR研究的不断深入,人们对常规PCR进行技术改进而衍生出了多种PCR技术,如荧光定量PCR、逆转录PCR、RACE-PCR、巢式PCR、反向PCR、降落PCR等。
荧光定量PCR是在反应体系中加荧光基团,利用荧光信号实时检测PCR进程并通过标准曲线对初始模板进行定量分析。
逆转录PCR是以RNA为模板,首先利用逆转录酶反转录成cDNA,再以cDNA为模板进行PCR扩增。
RACE即cDNA末端快速扩增技术,利用RACE可以通过已知的部分cDNA序列来得到完整的cDNA的5′和3’端。
巢式PCR是利用两套引物进行两轮PCR扩增。使用巢式PCR进行连续两轮扩增可以提高扩增的特异性和灵敏度。
反向PCR是用反向的互补引物来扩增两引物以外的未知序列的片段(常规PCR是对两引物之间的片段进行扩增)。
降落PCR是对PCR反应体系中退火温度进行优化,通过设置一系列不断降低的退火温度以提高PCR反应的特异性。
PCR技术在基因组学、分子生物学、临床医疗诊断及法医学等领域有着广泛的应用。下面为您介绍部分PCR应用领域:
逆转录是指以RNA为模板合成与其互补的cDNA的过程。逆转录PCR是将RNA的逆转录(reverse transcription) 和cDNA的聚合酶链式反应(PCR)相结合的技术,故逆转录称为RT-PCR或反转录PCR。逆转录PCR实验原理是,提取组织或细胞中的总RNA,以RNA为模板,利用逆转录酶反转录成cDNA,再以cDNA链为模板进行PCR扩增,从而获得大量拷贝。逆转录PCR的出现使RNA检测的灵敏性提高了几个数量级,使一些极为微量RNA样品分析成为可能。
逆转录PCR的用途广泛,可用于分析基因的转录产物、检测细胞中RNA病毒的含量、合成cDNA探针、直接克隆特定基因的cDNA序列等。如在临床上RT-PCR可以用于遗传病诊断、癌症检测、检测病人标本中的RNA病毒,如HAV、HCR、HIV等。在植物方面RT-PCR常用于研究环境胁迫对植物基因表达的影响,以及在特定的环境或生长阶段中植物体不同部位基因表达的差异性。使用RT-PCR检测分析RNA转录产物具有以下突出的优点:
逆转录PCR的模板是RNA,可以是总RNA、mRNA或体外转录的RNA产物。无论使用何种RNA,都需确保RNA中无RNA酶和基因组DNA的污染。
可以利用试剂盒从细胞(或组织)中提取得到RNA。模板RNA的纯度和完整性对于扩增的结果有很大影响,从细胞中分离RNA应注意尽量减少RNA酶的污染(RNA酶分布广泛,除细胞内源性RNA酶外环境中也存在大量RNA酶),在提取RNA时,应尽量创造一个无RNA酶的环境:避免RNA酶污染包括去除外源性RNA酶污染和抑制内源性RNA酶活性,主要是采用焦碳酸二乙酯(DEPC)去除外源性RNA酶,通过RNA酶的阻抑蛋白Rnasin和强力的蛋白质变性剂抑制内源性RNA酶。
用于反转录的引物有随机引物、通用引物(Oligo-dT)及基因特异性引物三种,下表为三种引物的介绍:
| 引物 | 适用范围 |
|---|---|
| 随机引物 | 适用于长的或具有发卡结构的RNA。适用于rRNA、mRNA、tRNA等所有RNA的反转录反应。主要用于单一模板的RT-PCR反应 |
| Oligo-dT | 适用于具有PolyA尾巴的RNA(原核生物的RNA、真核生物的rRNA和tRNA不具有PolyA尾巴) |
| 基因特异性引物 | 与模板序列互补的引物,适用于目的序列已知的情况 |
引物最好选择Oligo-dT或基因特异性引物,随机引物会从RNA的多个位点(包括核糖体RNA)开始转录,特异性低。Oligo-dT引物同大多数真核细胞mRNA3’端的poly(A)尾杂交,与使用随机引物相比特异性高。但是Oligo-dT引物对RNA样品的质量要求较高,对于从福尔马林固定的组织中提取的劣质RNA不适合用Oligo-dT引物。
逆转录酶是存在于RNA病毒体内的依赖RNA的DNA聚合酶。RT-PCR实验中的逆转录酶需要具有以下二种活性:
在选择逆转录酶时,建议选择无RNaseH① 活性(RNaseH-)的逆转录酶。具有RNaseH活性的逆转录酶的RNaseH活性会与聚合酶活性竞争RNA模板与DNA引物(或cDNA延伸链)形成的杂合链,并降解杂合链中的RNA链。被RNaseH活性所降解的RNA模板不能再作为合成cDNA的有效底物,降低了cDNA合成的产量与长度。
从细胞材料中提取RNA→RNA加入到含有逆转录酶、引物、dNTPs的反应体系中→退火,引物与RNA链配对→延伸,逆转录酶合成互补cDNA链变性→常规PCR反应流程(变性、退火、延伸,如此多次循环)。
RT-PCR的实验操作分为“一步法”与“两步法”两种。一步法RT-PCR能克隆微量mRNA而不需构建cDNA文库(即cDNA合成与PCR反应在同一Buffer及酶中进行,一步法完成),省略了cDNA与PCR之间的过程。两步法RT-PCR首先用反转录酶合成cDNA,然后以cDNA为模板进行PCR,即RNA反转录与PCR扩增分两步进行。一步法与两步法具有以下区别:
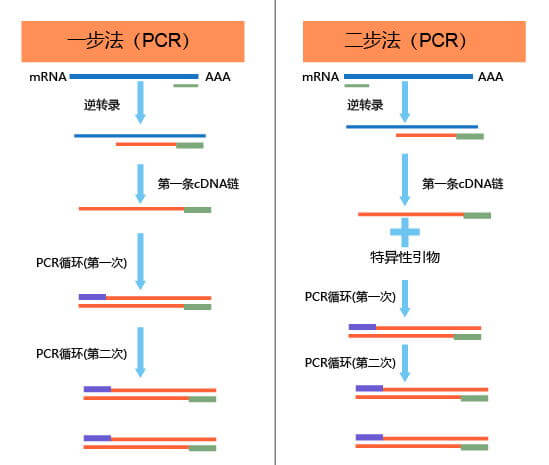
图2:“一步法”与“两步法”比较
注:
① RNase是RNA水解酶的统称,包含RNase A,RNase H等,RNase A可以水解单双链RNA,RNase H主要水解RNA与DNA杂交双链中的RNA
[1]孙晓东,王燕桑明.逆转录PCR(RT-PCR)实验操作要点[J].现代医药卫生,2009,25,13:2049-2049;
[2]陈余朋,张声,陈林莺.RT-PCR一步法与RT-PCR两步法比较[J].福建医科大学学报,2003,10,37:100-102;
[3]StephenA.Bustin.QuantificationofmRNAusingreal-timereversetranscriptionPCR(RT-PCR):Trendsandproblems[J].Biomarkers, 2005, 10(6):429-438;
[4]郭若霖,郭善一,鲍秋野等.竞争性RT-PCR测定法及BMP-2mRNA的定量检测[J].中国生物化学与分子生物学报,2000,16,5:680-683;
[5]赵晓,马会勤.葡萄果实发育后期半定量RT-PCR内参基因的优选[J],中国农业大学学报,2010,15,3:7-14;
RACE即cDNA末端快速扩增技术(rapid amplification of cDNA ends),是一种基于逆转录PCR从样本中快速扩增cDNA的5′端及3′端的技术,由Frohman等人于1988年发明。利用RACE可以通过已知的部分cDNA序列来得到完整的cDNA的5′和3′端。RACE的特点是在仅已知单侧序列可供设计特异性引物时,应用RACE技术仍能完成扩增,因此RACE技术也成为单侧PCR。
RACE包括3′RACE和5′RACE,分别用于cDNA3′端和5′端的扩增。根据已知序列设计特异性引物,利用3′RACE获得3′端序列(基因特异性引物→3′末端),利用5′RACE获得5′端序列(基因特异性引物→5′末端),最终获得完整的cDNA序列。3′RACE与5′RACE原理有所不同,下面分别做一介绍。
RACE的实验样本包括总RNA,poly(A)+RNA等。首先根据mRNA3′末端天然存在的Poly(A)尾部设计反转录引物逆转录获得第一条cDNA链。根据已知的cDNA序列设计基因特异性引物(gene specific primer,GSP)合成第二条cDNA链。随后以基因特异性引物(GSP)及正义链3′末端引物作为一对引物,对得到的cDNA链进行PCR扩增,从而得到cDNA的3′端序列(基因特异性引物→3′末端)。


根据已知的cDNA序列设计基因特异引物(GSP),逆转录获得第一条cDNA链,同时用末端脱氧核苷酸转移酶(TdT)在cDNA3′端加Poly(C)尾。依据Poly(C)尾设计特定引物合成第二条cDNA链。随后以第二条cDNA链为模板利用基因特异性引物合成双链cDNA。最后以基因特异性引物(GSP)及反义链3′末端引物为一对引物进行PCR扩增获得cDNA5′端序列(基因特异性引物→5′末端)。
在实际操作中,为了提高结果的特异性实际操作步骤会比上述原理复杂,此处只做原理性讲解,具体提高特异性的方法下文会有描述。
由于某些mRNA模板过程或模板二级结构含量过高,仅利用poly(A)尾来获得全长cDNA比较困难,甚至无法实现。因为长的mRNA(或二级结构含量过高)的反转录过程往往会提前终止,导致合成的cDNA第一链不完整,最终产生大量的非特异性产物。同时实验操作繁琐,在RACE技术出现之前,获得mRNA的全长反转录物cDNA往往需要数周甚至数月才可以完成。RACE技术为cDNA克隆方法开辟了新纪元,使用RACE技术可在1-2天内获得cDNA全长序列,同时特异性大大提高。
RACE技术相对于其它克隆全长cDNA的方法(如转座子标签技术、图谱克隆技术、mRNA差异显示技术等)具有价廉、简单和快速等特点。用RACE获得cDNA克隆只需几天的时间,而且对低丰度的起始反应物质,也能迅速反馈是否有目的产物生成。RACE所使用的起始总RNA或mRNA仅需ng级别,可扩增出丰度低于0.00001%的RNA样本,甚至仅有几个RNA分子亦可被检测出来。
尽管RACE的方法很有实用价值,但是要成功的应用该技术还是比较困难的,尤其是5′RACE,反转录、加尾、PCR这三个连续的酶促反应任一步骤的操作失误都会引起实验的失败,即使酶促反应步骤能顺利进行,也有可能会产生大量的非特异性产物。要做好RACE实验并不容易,实际操作中存在不少困难,需要采取多种措施以提高产物的特异性。
传统RACE技术存在诸多缺点,其特异性低,产物可能是单一产物、多个产物,甚至是不能分辨的连续条带。为了提高扩增的特异性,需要在传统RACE技术基础上进行改进。RACE技术的改进主要涉及引物的设计及PCR技术的改进两部分。
随着分子生物学技术的发展,科学家结合其它的分子生物学技术对最初的RACE技术进行了改进,从而丰富了RACR技术的类型。目前使用的RACE技术包括:经典RACE、Adapter-Ligated RACE、RLM-RACE、Cap-switching RACE和环形RACE等等。
Adapter-Ligated RACE是Adapter-Ligated PCR技术与RACE技术的结合。利用T4连接酶将接头与cDNA两末端连接,在PCR循环的退火步骤中,由于短cDNA退火温度低,两端接头容易发生退火,形成锅柄状结构,两端接头结合阻止引物与模板结合,终止PCR反应。长cDNA的退火温度高,不易形成锅柄结构,因此引物可以与接头结合,实现延伸。Adapter-Ligated RACE可以让长片段cDNA的克隆在扩增反应中占主导,从而尽可能多地得到目的基因的序列信息。
利用断裂的mRNA5′端没有帽子结构的特点,事先加入牛小肠碱性磷酸酶(CAP)将断裂mRNA5′末端暴露的磷酸基团切除。再加入烟草酸焦磷酸酶(TAP),TAP具有切除mRNA帽子结构的催化活性,能够使mRNA5′端暴露一个磷酸基团,接着在T4连接酶的催化下将衔接头与经过活化的mRNA5′端链接。经过钝化的mRNA是不能与衔接头链接的。经过这样处理后,便可以扩增目的mRNA5′端片段。
第一步以poly(T)作为引物对mRNA3′端克隆。当新和成cDNA延伸到mRNA5′帽子结构时,加入莫洛尼鼠白血病病毒逆转录酶(MMLV)在cDNA3′端加入若干胞嘧啶poly(C)。MMLV所催化的加尾反应需要依赖模板和帽子结构的存在,因此只有完整的cDNA末端才会加上胞嘧啶残基,接着加入特异性引物,该引物在3′末端含有含有多聚鸟嘌呤核苷酸poly(G)可与cDNA末端新添加的多聚胞嘧啶核苷酸poly(C)退火,在DNA聚合酶的催化下以新添加的引物为模板实现接头转化,从而向cDNA3′端引入特异性序列,最后再进行PCR。
环形RAC利用poly(T)引物进行逆转录PCR反应扩增第一条cDNA。经RNaseH降解模板后加入T4链接酶,加入T4连接酶时会发生环化反应或串联反应。无论是环化反应或是串联反应的产物都可以根据已知序列设计新引物来补充第二条链。环状分子或串联分子产生第二条cDNA链后,在未知区域的两侧设计一对引物将未知区域置换到已知序列中间,进行普通PCR。
需要说明的是,RACE技术种类繁多,但目前没有任何一种RACE技术能完美地扩增多有类型的RNA,每一种RACE技术都有其适合扩增的RNA种类,比如经典RACE适合扩增有poly(A)尾结构的RNA,Cap-switching RACE技术适合于扩增具有5′端帽子结构的RNA。
将RACE扩增得到的cDNA片段克隆至载体中,使用仪器对克隆后的质粒进行测序,最终得到该cDNA的序列。德泰生物提供杂交瘤细胞测序服务,可对抗体的可变区、重链、轻链及全长进行测序。
[1]. 徐烨,刘雅婷等.几种主要的RACE技术及应用[J].中国农业科技导报,2012,14(2):81-78;
[2]. 邓雪柯,殷建华等.3中5′RACE技术的比较与优化[J].成都医学院院报,2007,2,1:20-23;
[3]. 陈启龙.RACE技术的研究进展及其应用[J].2006,6,3:95-98;
常规PCR在对模板进行扩增的过程中,引物与模板之间会出现非特异性配对,导致产生非特异性产物。为了提高PCR扩增的特异性,人们对常规PCR进行技术改良,发明了巢式PCR(nested PCR)。
巢式PCR是指利用两套PCR引物进行两轮PCR扩增,第二轮的扩增产物才是目的基因片段。巢式PCR的实验原理为根据DNA模板序列设计两对引物,利用第一对引物(称为外引物)对靶DNA进行15-30个循环的标准扩增(见图1);第一轮扩增结束后将一小部分起始扩增产物稀释100-1000倍加入到第二轮扩增体系中作为模板,利用第二对引物(称为内引物或巢式引物,结合在第一轮PCR产物的内部)进行15-30个循环的扩增(见图1),第二轮PCR的扩增片段短于第一轮。

图1:巢式PCR扩增流程
与常规PCR技术相比,两套引物的使用提高了扩增的特异性,因为和两套引物都互补的靶序列很少。如果第一次扩增产生了错误片段,内引物与错误片段配对扩增的概率极低,因此提高了PCR扩增反应的特异性与灵敏度。
巢式PCR的注意事项与常规PCR基本相同。除此之外,巢式PCR实验还需要注意两轮扩增引物的比例:如果第一次引物过量的话,剩余引物第二次PCR扩增的时候同样能有一定的产量,这对于第二次PCR反应而言就是非特异性的产物。在第一次PCR时,应尽量摸索引物最低的加入量,同时适当增加循环次数,尽量消耗体系中的残余引物。
除了常规巢式PCR外,巢式PCR还有多种形式的扩展,下面分别做一介绍:
巢式PCR是利用第一轮PCR产物作为第二轮PCR的模板,除使用第一轮的一对特异引物之外,在第二轮PCR反应中使用一对新的特异引物,共使用四条特异性引物进行DNA扩增。半巢式PCR与巢式PCR原理相同,只是在第二轮PCR反应中使用的引物有一条为第一轮PCR的引物,这种利用三条引物进行两次PCR扩增的方法称为半巢式PCR( semi-nested PCR)。
在一些需要利用巢式PCR进行扩增的实验中,如果基因的3’末端或者5’末端无法设计出两条引物,可以使用半巢式PCR。
反转录巢式PCR( RT-nested PCR)是在反转录PCR的基础上发展起来的,在通过反转录获得cDNA的基础上,对目的基因进行巢式PCR扩增。它和简单的反转录PCR一样是用于检测某种RNA是否被表达或者比较其相对表达水平,但是特异性更高、可靠性更强,可用于拷贝数较低的RNA的扩增,例如扩增丙型肝炎病毒(HCV)感染者体内的HCV基因。
单管巢式PCR是在传统巢式PCR的基础上将两对PCR引物作特殊的设计,巢式外侧两个引物为25bp,退火温度比较高(68℃);巢式内侧两个引物为17bp,退火温度较低(46℃)。通过控制退火温度(68℃)使外侧引物先行扩增,经过20~30次循环后(第一轮PCR结束),再降低退火温度(46℃)使内侧引物以第一次PCR产物为模板进行巢式扩增。单管巢式PCR两轮PCR反应均在一个PCR管中进行,减少了交叉污染的可能性。
共有序列巢式PCR( consensus nested PCR),又称为共有引物巢式PCR( consensus primer nested PCR),根据同一种属内较为保守的序列设计简并引物,通常第一轮PCR引物的简并碱基较多,第二轮PCR引物的简并碱基较少一些,扩增长度为200~300bp。引物通常设计在能够区分微生物的不同亚型的区域内。对于某一种生物,例如病毒,种属内型别很多,但检测样本中的病毒型别又不确定,使用共有序列巢式PCR扩增获得目的序列,进而通过测序获得未知微生物的信息,是一种敏感而又简便易行的检测方法。
共有序列巢式PCR引物设计尤为重要,在引物设计之前要搜集可能相关的所有DNA序列,利用软件进行严格的序列对比分析,从中找出保守的序列,在这部分序列中可能仍存在一些核苷酸的多样性,则在具有多样性核苷酸的位置上设计为简并碱基,将所出现的所有核苷酸多样性均要考虑,第二轮PCR扩增产物长度控制在200~300bp左右。由于引物中简并碱基较多,要摸索适合的退火温度。
巢式PCR大多应用在当模板DNA含量较低时,用一次PCR难以得到满意的结果,这时用巢式PCR的两轮扩增可以得到很好的效果。
巢式PCR操作简单,所需条件与常规PCR相同,但与常规PCR相比进一步提高了反应的特异性与灵敏性,在微生物学、生物信息学、生物医学等方面有着极其广泛的应用。例如巢式PCR技术与RFLP(限制性片段长度多态性)技术结合,通过设计高度保守序列的引物对待检物种DNA进行PCR扩增,对PCR产物进行RFLP分析,从而完成DNA分子水平上的多态性检测,该法常用于流行病学的调查和临床常规检测。
[1]程小华,杨飞等.巢式PCR检测血清样品中HCV RNA的稳定性[J].2011,32(4):526-528;
[2]黄留玉.PCR最新技术原理、方法及应用[M].北京:化学出版社,2011:38-51;






南京德泰生物工程有限公司 Nanjing Detai Bioengineering Co.,Ltd. ©2025 All Rights Reserved
 苏公网安备32011202001300
苏公网安备32011202001300